
Applying Human Performance Models In FDA-Regulated Environments
By Joanna Gallant, owner/president, Joanna Gallant Training Associates, LLC

After giving a recent course on reacting to human error, one of the attendees sent me a variety of questions about how to apply the human performance models we discussed in FDA-regulated environments. Answering the questions was a good exercise in melding the two sets of concepts, and became something worth sharing more widely — in the form of a two-part, Q&A-style article. This article will provide regulatory context, address confidentiality concerns, and walk through an application based on an actual FDA 483. Part 2 will explain how to accurately identify human errors, determine when a deviation or nonconformance requires CAPA, and get started using human performance improvement tools and processes in your organization.
How do regulators identify and approach human performance problems?
Human performance problems regularly appear as citings in FDA 483s and warning letters. Many highlight data integrity or aseptic/cleanroom performance problems and the related behaviors observed by FDA investigators — like this example of a cleanroom behavior observation from a 2011 483:
During aseptic filling operations, we observed an operator resting his gloved hands on his midsection near the gown zipper. Per our request, the operator was counseled by his supervisor regarding proper aseptic technique during our tour. However, following the discussion with his supervisor, the operator resumed the practice of resting his gloved hands on his midsection near the gown zipper. We observed this operator performing and assisting in multiple line interventions during our tour. Your firm’s current aseptic techniques SOP lacks instructions for maintaining distance between gloved hands and sterile gowning during filling operations.
The observed human behavior and performance problems have been linked to a variety of regulatory references, including citations of aseptic processing area failures, failures to thoroughly investigate deviations or out-of-specification (OOS) results, failures to maintain complete laboratory testing data, and more.
I had an opportunity to speak with a current FDA investigator and former IMB (Irish Medicines Board) investigator, and asked about their approach to identifying and addressing possible human error/human performance concerns. At the time, I wanted to understand why training was so frequently cited when the source of the error or performance issue may lie elsewhere. I came away from the conversations understanding this as the chain of thinking:
- First, did the error result from a process failure? (Was there a problem or gap in the process or procedures?)
- If it’s not a process failure, then did it result from a training failure? (Was the training not effective? Did the person not know how to do the job?)
- If it’s not a process or training failure, did the error result from an oversight failure? (Was the person’s performance not appropriately managed/guided?)
- If it’s not related to the process, training, or oversight, then it could be a human error.
While citings of process and training issues/failures still appear, several recent data integrity observations cite failures of management to oversee or control the process — pushing further down into this logical cause chain. As an example, consider this excerpt from an October 2015 warning letter:
According to your responses … your manufacturing staff did not exhibit acceptable documentation practices, and your chemist or microbiologist each neglected his work. However, your management is responsible for routine oversight of manufacturing and testing operations, including the activities of operators and other personnel, and your responses do not address the failure of management and the flaws in your overall quality system.
In response to this letter, conduct and provide the results of a comprehensive investigation into your poor documentation practices. Your investigation should address the flaws in your quality systems and management oversight that led to these serious deficiencies. … Also provide your procedures for addressing deviations from acceptable documentation practices, including training and oversight of personnel whose duties require preparation and review of API records.
Regulatory expectations for management responsibilities, which include process oversight, monitoring process performance, committing appropriate resources, communicating expectations, and ensuring that personnel are appropriately qualified for their positions, are laid out in ICH Q10 and the FDA’s quality system guidance.
These elements fit into Gilbert’s Behavioral Engineering Model (BEM) (Figure 1). The model identifies the elements that influence human behavior (performance), where desired behavior is the output of the combination of these elements.
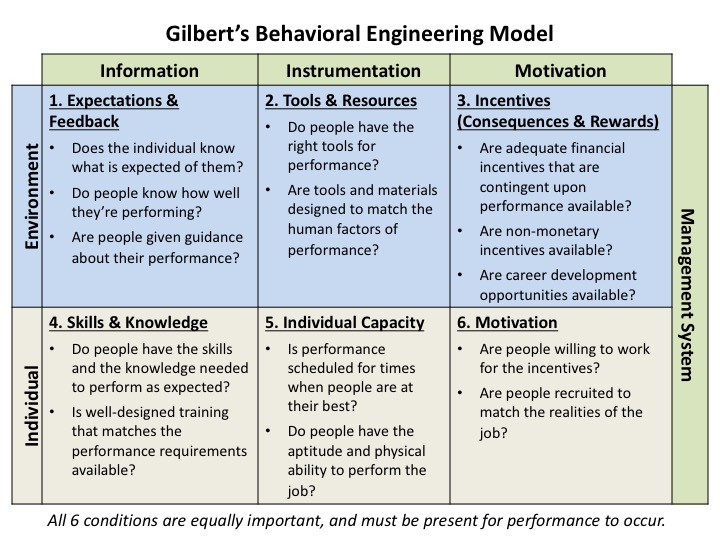 Figure 1: Gilbert's Behavioral Engineering Model (adapted from Deb Wagner's HPT Toolkit)
Figure 1: Gilbert's Behavioral Engineering Model (adapted from Deb Wagner's HPT Toolkit)
The model identifies:
- The categories of elements that affect/drive human performance
- Which elements must be provided in or by the work environment
- Which elements the individual brings to the task
- And that the management system must monitor/drive these elements to ensure desired performance is achieved
Conversely, then, performance issues are caused by deficiencies in one or more of these elements. Many, if not all, of these elements have been identified as contributing to human performance issues observed in agency inspections, including in the two examples provided earlier.
But aren’t performance-related elements confidential? Can we really use these tools/models in deviation/nonconformance (NC) investigations?
Some performance information (like performance reviews, salaries, ratings, etc.) is, and should be, confidential. This information has no bearing on, and should not be included in, the investigation process.
However, when a human performance issue potentially impacts product or process quality, that risk to quality must be addressed. The process of investigating and remediating quality issues should not require confidentiality. Some outputs of the process could fall into the confidential information category, but those likely don’t need to be included in the investigation record.
For example, the earlier 483 observation involves a performance issue that is clearly a GMP failure and product quality risk. It was cited as a “failure to establish and follow procedures to prevent microbiological contamination” against 21 CFR 211.113, a regulatory/legal requirement. This situation must be addressed because it’s a legal liability to the company — it’s a violation of an established regulation.
To address the observation, we would investigate the situation with a process that includes:
- Identifying the problem
- Determining the contributing and root causes
- Identifying appropriate CAPAs
- Implementing those CAPAs
- Checking the effectiveness of the CAPAs
The International Society for Performance Improvement (ISPI) Performance Improvement/Human Performance Technology (HPT) Model (Figure 2) also defines an investigation and remediation process.
{kind=link}
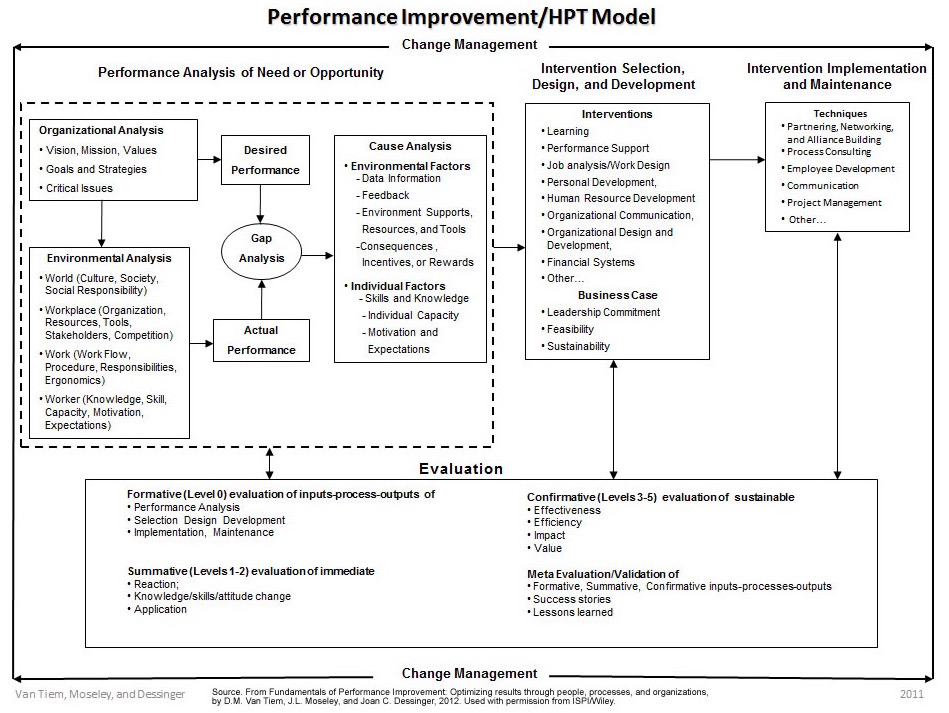
Figure 2: ISPI Performance Improvement/Human Performance Technology Model
At first glance, it appears very different from the investigation process above. In reality, it simply points the investigation to personnel/behavioral topics.
The table below shows how the two investigation models align.
|
GMP Investigation Model Element |
Equivalent HPT Model Element |
|
Problem identification |
Gap analysis |
|
Determine contributing and root causes |
Cause analysis (note the similarities between this and the BEM) |
|
CAPA identification |
Intervention selection, design, and development |
|
CAPA implementation |
Intervention implementation and maintenance |
|
CAPA effectiveness check |
Evaluation |
Table 1: Comparison of GMP and HPT investigation models
Continuing with our example, if we look into this situation, based on what is presented in the 483, here’s what we’d likely do/find:
The Problem/Gap Analysis
An operator exhibited inappropriate aseptic behaviors in a cleanroom during processing.
Cause Analysis
The observation states that an aseptic technique SOP was in place, but was missing a piece of information directly related to the operator’s behavior (which links to BEM element #2). We’ll also make a few assumptions about the situation:
- The involved operator was gowned appropriately. (The observation doesn’t reference incorrect/inappropriate gowning — only behavior while gowned.)
- Only the involved operator’s behavior is in question. (The observation focuses on one person — any others present at the time of the observation were not mentioned.)
- It then follows that operators had received training (links to BEM element #4) on both gowning and cleanroom behaviors, even though the SOP was missing information. (No other operator exhibited the same behavior during the observation.)
Our root cause analysis (RCA), using 5 Whys, might then go like this:
- Why was only one operator exhibiting incorrect behavior? It’s possible the operator had not received feedback that his performance is not meeting expectations (links to BEM element #1).
- Then why did the operator continue the incorrect behavior after being corrected? It’s possible that there are no consequences to poor behavior (links to BEM element #3), and therefore the operator has no motivation to change behavior (links to BEM element #6).
These questions lead us to a root cause of “Management/policy not enforced” (see my earlier article Identifying cGMP Issues Caused By "Management Problems" for management root cause categories). They’ve also identified that several elements within the Behavioral Engineering Model may not be sufficient to support the desired behavior.
CAPA Identification/Implementation, Intervention Selection/Implementation
While the observation cited a shortcoming in the procedure, simply revising the procedure would not fully address the situation or the observation, because it doesn’t address the lack of ongoing performance oversight or consequences for failures.
To address this observation, appropriate CAPAs would likely include:
- Updating the procedure to specify that gloves are not allowed to contact gowning, and add any other missing content, then training operators on the revised procedure — which strengthens the procedure and provides performance support (BEM #2).
- Ensuring a well-designed training/qualification process for aseptic techniques is in place, and that the new procedural expectations are factored into the process — which ensures the training provided matches the task expectations. (BEM #3)
- Establishing a disqualification process that rescinds an individual’s authorization to perform tasks when evidence of incorrect performance exists — which institutes consequences for inappropriate performance of the task. (BEM #3)
- Disqualifying the involved operator from entering a cleanroom and performing aseptic techniques/tasks until they complete a requalification process in which they demonstrate proper behaviors/techniques — which provides feedback to the operator that they are not meeting expectations. (BEM #1)
- Completing an observational performance assessment of other cleanroom operators to verify appropriate behaviors/techniques, and disqualifying/requalifying anyone who does not display appropriate behaviors/techniques — which provides feedback to remaining operators on the appropriateness of their performance (BEM #1)
- Establishing a periodic requalification process that includes criteria to assess personnel behaviors — which ensures a periodic check of performance is done. (BEM #1)
- Requiring area management to perform and document periodic on-floor observations that enable disqualification of operators who do not meet performance expectations. The on-floor observations become part of a procedure with specific documentation to complete as evidence of ongoing performance. This ensures frequent operational oversight occurs. (BEM #1)
CAPA Effectiveness/Evaluation
On an ongoing basis, the qualification, disqualification, requalification, and on-floor oversight processes should provide evidence that operators possess the skills/ability to operate in a cleanroom, while weeding out those who exhibit inappropriate behaviors — which are now defined in the aseptic techniques SOP.
So now, from the GMP standpoint, only qualified personnel are authorized to perform processes. Management can — and does — identify and exclude those who would compromise the process through inappropriate behaviors. And, the company meets regulatory requirements for the process.
The investigation and CAPAs relate back to fulfilling the defined legal/regulatory obligations, as well as to addressing the gaps identified in the BEM. Nothing in this investigation leads into confidential HR-information territory.
While further performance management actions (such as individual performance reviews reflecting poor performance, holding the operator/area management accountable for poor performance, or possible reassignment of job roles due to performance issues) may occur, they do not affect the investigation/remediation process, and occur outside of the GMP process, where confidentiality can be maintained.
With this as a backdrop, Part 2 will seek to answer these questions: Can a failure actually be attributable to human error? Can we really say in a deviation or NC that a well-trained employee had a “slip” or a “lapse”? Does every deviation/NC demand an investigation and corrective action (even the low-risk ones)? How can companies get started with applying human performance tools and processes to improve performance?
Additional References
- Gilbert, T. F. (1978). Human Competence: Engineering Worthy Performance. New York: McGraw-Hill.
- Binder C. (1998). The Six BoxesTM: A Descendent of Gilbert's Behavior Engineering Model. Performance Improvement, 37(6), July/August 1998, 48-52. Retrieved 22Mar2016 from http://www.binder-riha.com/sixboxes.pdf