Classification And FDA Regulations: Medical Device, Pharma, and Combination Products
By Oliver Stauffer

Package integrity testing is regulated by the FDA and risk assessments determine the extent to which testing needs to occur. How the FDA regulates various product classes and the guidance documents driving regulatory enforcement varies from product to product. An important distinction is the difference between medical device, pharmaceutical products, and those considered combination products that ride the divide.
 The FDA recognizes a medical device as any instrument, apparatus, machine, implant or other similar article which does not achieve its primary intended purpose through chemical action (purely a device). The moment the device includes a chemical component that actively reacts during its intended use, it becomes a combination product. A heart stent that is drug emitting is a combination product. A syringe without the liquid in it (just the components) is a device, and the moment it is designed to hold liquid, such as a pre-filled syringe, it becomes a combination product.
The FDA recognizes a medical device as any instrument, apparatus, machine, implant or other similar article which does not achieve its primary intended purpose through chemical action (purely a device). The moment the device includes a chemical component that actively reacts during its intended use, it becomes a combination product. A heart stent that is drug emitting is a combination product. A syringe without the liquid in it (just the components) is a device, and the moment it is designed to hold liquid, such as a pre-filled syringe, it becomes a combination product.
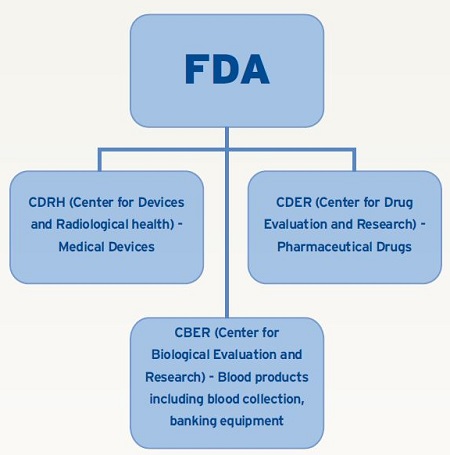
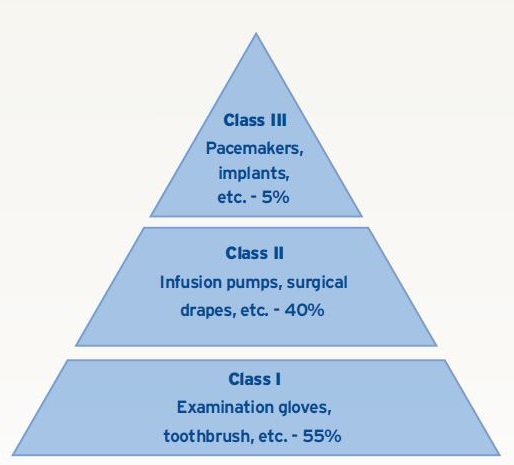
CDRH is the division of FDA that regulates medical devices. Medical devices are divided into three classes, ranging from a tooth brush (Class I) to a knee implant (Class III). FDA defines Class III devices as those that support or sustain human life or are of substantial importance in preventing impairment of human health. Class III devices only account for 5% of the medical device products in the field, but certainly consume the majority of attention by the FDA with focus on better inspection technologies than Class I or II.
ISO Standard 11607 highlights FDA Consensus Standards that the FDA deems acceptable in assuring package quality for medical devices. These Standards are essentially guidance documents. All FDA Consensus Standards are ASTM test methods, with vacuum decay (ASTM F2338) being one of them.
Treatments that do involve a chemical reaction, such as a pharmaceutical tablet or parenteral product, are regulated by either the CBER or CDER divisions of the FDA. These products also vary in terms of risk to the patient, ranging from oral dose to injection. CBER and CDER do not prescribe to ISO 11607 as a guidance document and ASTM test methodology carries less weight. CBER and CDER rely on United States Pharmacopeia (USP) Chapter <1207> to provide guidance on appropriate inspection test methodology to assure package integrity. The newly updated and revised USP <1207> is a major shift that impacts how the FDA looks at package integrity testing. It’s important to learn about this newly revised chapter.