
FDA FY2019 Warning Letter Trends: A Closer Look At Drug Product Manufacturers
By Barbara Unger, Unger Consulting Inc.
This is the second article in a two-part series reviewing the FDA’s GMP drug warning letters from FY2019. Part 1 provided a high-level overview of the FY2019 data, including trends since FY2013, where this year we saw a dramatic increase in warning letters issued to sites in the U.S., far exceeding the number issued to sites outside the U.S. for the first time since before FY2013. In Part 2, we take a deeper dive into the types of drug product manufacturers that received warning letters and their locations. This includes over-the-counter (OTC) drug product manufacturers and homeopathic drug product manufacturers, human cell therapy product manufacturers, firms associated with the nitrosamine contamination of angiotensin II receptor blockers (ARBs), and firms that repackage APIs. We finish up with some interesting data on how frequently the FDA recommends that firms hire qualified consultants, both inside and outside the U.S., and the FDA’s request for “independent assessments” in warning letters.
provided a high-level overview of the FY2019 data, including trends since FY2013, where this year we saw a dramatic increase in warning letters issued to sites in the U.S., far exceeding the number issued to sites outside the U.S. for the first time since before FY2013. In Part 2, we take a deeper dive into the types of drug product manufacturers that received warning letters and their locations. This includes over-the-counter (OTC) drug product manufacturers and homeopathic drug product manufacturers, human cell therapy product manufacturers, firms associated with the nitrosamine contamination of angiotensin II receptor blockers (ARBs), and firms that repackage APIs. We finish up with some interesting data on how frequently the FDA recommends that firms hire qualified consultants, both inside and outside the U.S., and the FDA’s request for “independent assessments” in warning letters.
The term “compounding pharmacy,” as used here, includes outsourcing facilities and for the purpose of this analysis is considered a separate category from other drug manufacturers based on their legal foundation. These sites are located in the United States, but they are not considered with data from U.S. drug manufacturing sites in most analyses in this article. Outsourcing facilities were established by the FDA under an amendment to the Food Drug & Cosmetic Act in November of 2013. The data from these sites is included in Figure 3. All other figures omit this market segment data. All data reflect fiscal years, with the exception of Figure 2, which shows the trend over time of issuance of warning letters to OTC firms in calendar years (CY).
Drug Product Manufacturers
Let’s take a deeper dive into drug product manufacturers that were the recipients of warning letters in FY2019. Drug product manufacturers are a diverse group, including firms that manufacture OTC/homeopathic drugs, manufacturers of human cell and tissue products, and prescription dosage form manufacturers of biotech or chemically synthesized sterile products and non-sterile products.
OTC/Homeopathic Drug Products
Figure 1 shows that OTC/homoeopathic drug product manufacturers received more than half of the drug product warning letters in FY2019. This continues a trend that started a few years ago. Figure 2 shows that over half of these firms that received warning letters in FY2019 were in the U.S., with China and India a distant second and third. Twenty of the warning letters issued to OTC/homeopathic firms also included a data integrity component. Figure 3 shows trends for issuance of warning letters to these firms over calendar years. Between CY2010 and CY2013, the number of warning letters issued to these firms ranged between one and six. The dramatic increase began in CY2017 and continues through this year. Many of the violations noted in these warning letters identify fundamental failures to understand and comply with GMPs, including, but certainly not limited to:
- Failure to test incoming raw materials and components, including APIs, for identity. Rather, materials and components are accepted on the basis of the supplier’s certificate of analysis (CoA), although the supplier has not been qualified and the results on the CoA have not been validated and periodically confirmed.
- Drug product is released for distribution without adequate testing, including identity and strength of the active ingredient and adequate microbiological evaluation.
- Manufacturing processes are not validated, and the firm does not have a process for ongoing monitoring to ensure the process remains in a state of control.
- Absence of basic SOPs for GMP activities, including, but not limited to, the responsibilities of the quality unit, change control, out of specification (OOS) investigations, deviation management, and complaint management, just to name a few
- Batch records did not have adequate detail of all the critical manufacturing operations.
- Inadequacy in water (likely purified water USP) generation and distribution systems, maintenance, sampling, and control
- And frequently, all of these shortcomings are combined into a statement that the quality unit does not have the appropriate power and authority to ensure that drugs have the quality, purity, potency, and safety that they are purported to have.
The types of deficiencies at these firms clearly skew any metrics that may be done on inspection observations and warning letter deficiencies. This is an important class of manufacturers, if only because of the number of individuals their products touch. We likely all have medicine cabinets with at least a few of these products that we use frequently. We probably have not seen the end of the FDA’s laserlike focus on this product category, and I look for more in FY2020.
Human Cell Therapy Drug Products
We previously covered the FY2018 and 2019 inspection observations for human tissue and cell therapy products. This is a diverse product category that includes sites such as fertility clinics, stem cell clinics, and gene and cell therapy manufacturers. This group received 10 warning letters in FY2019. Four of them were sent to fertility clinics and six were issued to firms selling either cord blood products (Stemell, Cord for Life, and Genetech), an amniotic membrane patch allograft (Stratus Biosystems), or adipose tissue harvested and modified for autologous use (StemGenex Biological Laboratories) and advanced stem cell allografts (Human Biologics of Texas/Globus Medical). Genetech, which received its warning letter on Nov. 19, 2018, distributed its products to Liveyon LLC. Liveyon Labs Inc. in Yorba Linda, CA, received a warning letter early in FY2020, on Dec. 5, 2019. Liveyon Labs initiated a Class II recall of 91 lots of HPVC cord blood products on Sept. 28, 2018 and terminated the recall on Nov. 5, 2019. The FDA gives the reason for the recalls as “Human tissue allografts, where in-process controls were not followed allowing tissue to be processed in a manner that could cause contamination or cross-contamination during processing, as well as the introduction, transmission or spread of communicable disease through the use of the tissue, were distributed.
Stem cell therapies have been problematic, with many firms operating outside the FDA’s framework for regenerative medicines. On June 4, 2019, the courts held that U.S. Stem Cell Inc. of Florida and its chief scientific officer “…adulterated and misbranded a stem cell drug product made from a patient’s adipose tissue.” The firm received a warning letter in 2017 for marketing a stem cell product without FDA approval and for significant non-compliance with GMPs. This is clearly a problematic area for the FDA, and we will watch carefully as the FDA’s enforcement discretion identified in a final 2017 guidance draws to a close late in 2020. I expect letters to human cell and tissue firms to increase in FY2020 and beyond as both gene and cell therapy firms expand in number and enforcement discretion is slated to end for the hundreds of rogue stem cell clinics. On the gene therapy side, the Pink Sheet (Jan. 26, 2020) recently identified six product candidates currently under review at the FDA, with five designated as breakthrough therapies. A recent court case in California, however, calls into question the FDA’s approach to regulation of autologous cell therapy. More to come here, for sure.
Prescription Drug Products other than HCTPs, Parenteral, and non-sterile Dosage Forms
The remaining warning letters were issued to drug product manufacturers, including parenteral products and non-sterile drug products including solid oral dosage forms and creams/ointments. The FDA continues its focus on parenteral products, particularly those made using aseptic processing, as a category of highest risk products.
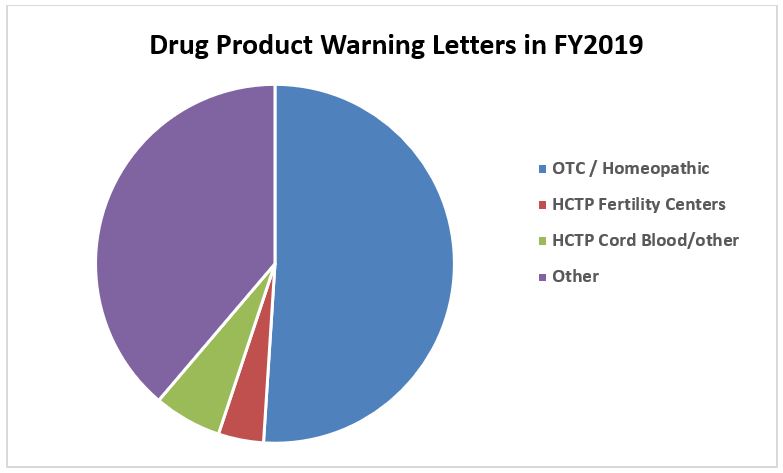
Figure 1: Drug product warning letters by product type
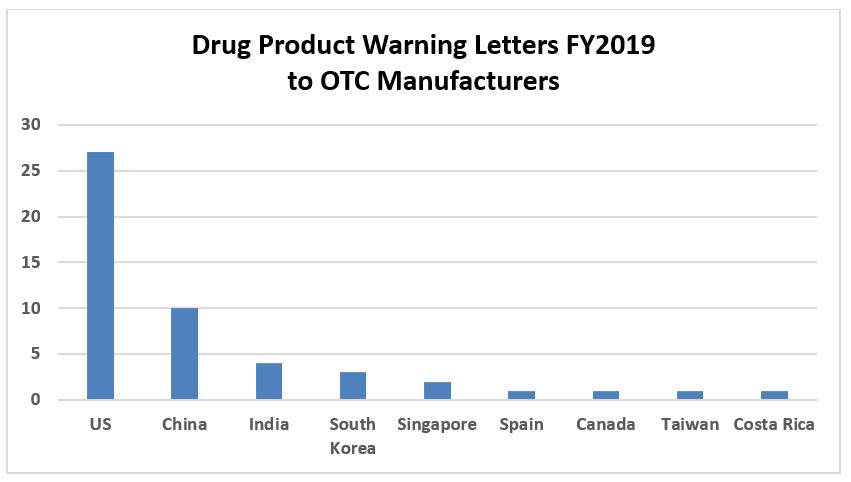
Figure 2: OTC warning letters by country

Figure 3: Warning letters issued to OTC Firms, CY2014-CY2019
Other Notable Topics In Warning Letters In FY2019
- The FDA generally identifies the information firms must provide in response to warning letters. This year, the frequency with which the FDA requests an “independent assessment” of the topics in question struck me. The FDA has clearly decided that many firms are not capable of performing an effective assessment and identifying appropriate remediation activities.
- In 76 warning letters, the FDA either suggests the firm hire qualified consultants or acknowledge it has done so, and sometimes both. Seventy-nine percent of warning letters issued OUS and 62 percent of the warning letters issued for firms located in the U.S. included this recommendation.
- The identification of the various nitrosamine contaminants in ARB API and drug products resulted in a collection of warning letters issued to the following firms, for example : Zhejiang Huahai Pharmaceutical Co. Ltd, Aurobindo Pharma Limited, Hetero Labs Limited, Lantech Pharmaceuticals Limited, Mylan Laboratories Limited, and Torrent Pharmaceuticals Limited. These sites manufacture either a “sartan” API and/or drug product. All of the sites to which these warning letters were issued are in China or India. Although the warning letter issued to Mylan Laboratories was sent to the CEO in the U.S., the site in question is in India. Among this collection, the one issued to Lantech Pharmaceuticals identified the problematic issue of recovered solvents and the lack of control of these solutions, which is now considered to be one source of nitrosamine contaminants. In addition to warning letters, dozens of products have been the subject of Class II recalls. The FDA has published methods for identification and quantitation of specific nitrosamine contaminants, along with interim acceptable daily limits. The EMA also addressed this topic and published Information on nitrosamines for marketing authorization holders on Sept. 19, 2019. On Oct. 9, 2019, the EMA published a “Questions and answers on Information on nitrosamines for marketing authorization holders.” WHO also published information on nitrosamine impurities.
- In FY2019, the FDA issued the first warning letter that cites failure to comply with requirements of the November, 2013 Drug Supply Chain Security Act (DSCSA) amendments to the FD&C Act. This warning letter was issued to McKesson Corporation in San Francisco on Feb. 7, 2019, based on observations from a form 483 issued at the close of the inspection on July 3, 2018. First of a kind enforcement actions are always of interest in terms of what they tell us about the FDA’s potential future enforcement in this area. Also, the text in the enforcement action provides insight into what the FDA found to be the specific objectionable activity or process, so others operating in this area can evaluate their procedures and processes.
The Feb. 7, 2019 warning letter identifies an inspection of the corporate headquarters in San Francisco, and it identifies three deficiencies where McKesson Corp failed to comply with the requirements of section 582(c)(4), identification of suspect product and notification. The warning letter describes three examples supporting the agency’s conclusions and three examples of inadequate corrective actions. Effective Jan. 1, 2015, trading partners are required to quarantine and investigate suspect product to determine whether it is illegitimate and to notify the FDA and trading partners if the illegitimate product is found. The FDA published guidance on the topic, Identification of Suspect Product and Notification, in December 2016. This was clearly a situation where the firm had plenty of time to come into compliance.
- Another interesting group of warning letters addresses the topic of information that repackagers include on the CoA provided to customers. The FDA places a high priority on ensuring a secure and accurate supply chain based in part on documentation accompanying purchased products, particularly certificates of analysis. Four warning letters were issued in FY2019 where repackagers did not identify the original manufacturer of the API, but rather transferred the information to their own letterhead and did not include any information regarding the original manufacturer. Three of the firms are in the U.S., the fourth in India. The three firms that received the four warning letters including this deficiency include: Vipor Chemical Privat Ltd., Spectrum Laboratories HERE and HERE, and B&B Pharmaceuticals Inc.
Spectrum Laboratories received two warning letters based on its practices at two sites. It attempted to convince the FDA that its practices were satisfactory, though that seems to have been woefully unsuccessful. The June 4, 2019 warning letter to Spectrum notes that because six of the repackaged APIs list only Spectrum’s name on the label, this falsely represents that Spectrum is the manufacturer. Thus, the FDA deems these APIs to be misbranded, because the labels are false and misleading. The FDA also notes that both of the sites had similar deficiencies and thus “…failures at multiple sites demonstrate that management oversight and control over the manufacture of drugs are inadequate.” Although the second Spectrum site received a warning letter for this same shortcoming, the FDA did not note the issue of misbranding.
Conclusion
FY2019 was a fascinating year for drug GMP warning letters in the diversity of topics addressed, depth of focus, and trends in enforcement actions. The most significant news is that the number of warning letters issued to sites in the U.S. increased significantly. For the first year since I started monitoring this in FY2013, warning letters issued to firms in the U.S. constituted a majority of the drug GMP warning letters, far outpacing India and China combined.
Warning letters issued in FY2019 were issued to a diverse group of firms both inside and outside the U.S. The number issued to OTC firms and HCT/P firms both increased. We saw a decrease, however, in the number of warning letters issued to compounding pharmacies/outsourcing facilities that began in FY2017. Similarly, we saw a decrease in the number of warning letters that identify data integrity shortcomings, again for the third year.
FDA issued a cluster of warning letters to firms associated with ARB APIs and drug products, including mention of the use of recovered solvents in at least two of them. Another group that received warning letters were repackagers that neglected to identify the original manufacturer of the product on CoAs. Along with these two groups, this year saw a focus on the API and drug product manufacturers for ARB. Enforcement for this group of products also included recalls of hundreds of lots of product. This year we also saw many warning letters where the FDA requested “independent assessments” from firms that received warning letters and recommended that many firms hire qualified GMP consultant(s) to assist them in coming into GMP compliance.
ForFY2020, I see a continuing focus on OTC manufacturers, as there are many of them and they seem to fail to understand the fundamentals of GMPs. The enforcement discretion with stem cell clinics is supposed to cease at the end of the year, but a court case in California could have significant impact on the FDA’s jurisdiction in the area of autologous stem cell clinics. At least six gene therapy products are currently under review at the FDA, so we will see those pre-approval inspections (PAIs), along with the start of routine inspections of the gene therapy firms that now have approved products. Sterile drug products, particularly those produced using aseptic processing will remain a priority. FY2020 could well be as interesting as FY2019.
About The Author:
 Barbara Unger formed Unger Consulting, Inc. to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including general GMP auditing, auditing gene and cell therapy manufacturers and suppliers, and auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Unger was the first chairperson of the Rx-360 Monitoring and Reporting work group that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She co-led the Rx-360 Data Integrity Working Group from 2017–2019. You can contact her at bwunger123@gmail.com.
Barbara Unger formed Unger Consulting, Inc. to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including general GMP auditing, auditing gene and cell therapy manufacturers and suppliers, and auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Unger was the first chairperson of the Rx-360 Monitoring and Reporting work group that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She co-led the Rx-360 Data Integrity Working Group from 2017–2019. You can contact her at bwunger123@gmail.com.