
FDA FY2018 Drug Inspection Observations And Trends
By Barbara Unger, Unger Consulting Inc.

 A comprehensive GMP intelligence program includes monitoring of health authority enforcement actions, including FDA Form 483s, Establishment Inspection Reports, warning letters, recalls, import alerts, consent decree agreements, and EU reports of GMDP noncompliance. This article presents the most recent publication of GMP drug inspection data from CDER, which addresses drug inspections conducted in FY2018. While the properties section of the document indicate it was created in December 2018, it was published on the FDA website in September 2019. We examine data from FY2018 and evaluate six years’ worth of trends in GMP inspection enforcement. The CDER drug inspection observations supplement the information previously published describing CDER Drug GMP warning letters issued in FY2018.
A comprehensive GMP intelligence program includes monitoring of health authority enforcement actions, including FDA Form 483s, Establishment Inspection Reports, warning letters, recalls, import alerts, consent decree agreements, and EU reports of GMDP noncompliance. This article presents the most recent publication of GMP drug inspection data from CDER, which addresses drug inspections conducted in FY2018. While the properties section of the document indicate it was created in December 2018, it was published on the FDA website in September 2019. We examine data from FY2018 and evaluate six years’ worth of trends in GMP inspection enforcement. The CDER drug inspection observations supplement the information previously published describing CDER Drug GMP warning letters issued in FY2018.
The presentation of data herein differs from data presented on the FDA website, even though it uses the same raw data. For example, I combine all observation listings that cite 21 CFR 211.192 into a single value, rather than identifying them in five separate line items with the exact same citation text. I also consolidate the more than 15 line items for §211.160(b) into a single line item. §211.88 (a) and (b) are consolidated into a single line rather than being divided into more than 20 line items. Similar consolidation is done for each of the §211 citations identified in Table 1. Consolidating the citations leads to slightly different conclusions. In my opinion, sometimes, slicing the observations so narrowly, as the FDA does, dilutes their impact.
The FDA’s data includes only Form 483s issued through its electronic system; it does not include Form 483s issued to API manufacturers, because §211 is not applied to those manufacturers, or Form 483s issued outside of the electronic system. Thus, the data does not represent the FDA's complete collection of inspection observations for the year. In past years, these data represented approximately one-third of all Form 483s issued, so conclusions must be tempered by the relative incompleteness of the data.
Executive Summary:
- The number of Form 483s included in this analysis remained reasonably consistent over the past five fiscal years, ranging between 645 and 716 inspections. (Table 1, row 2)
- The number of observations citing failures in §211.192 (investigation of discrepancies) was displaced from the top of this list in FY2018 for the first time since before FY2013.
- The two top citations in FY2018, §211.160(b) (Lab controls should include scientifically sound specifications) and §211.22(d) (Quality unit procedures shall be in writing and shall be followed) were previously second and fourth.
- Five specific §211 citations decreased markedly between FY2016 and FY2018.
FDA Form 483 Inspection Observations
Table 1 shows only the 15 most frequent inspection observation citations between FY2013 and FY 2018, while the tabulation on the FDA website shows all citations used in the fiscal year. The FDA uses the term “frequency” to represent the number of times the agency identified a specific citation in its tabulation. Table 1 presents those observations from the highest to lowest number for 2018, modified as described in the Introduction section of this article. Both Table 1 and Figure 1 show consistency in the years between FY2013 and FY2017 with respect to the identity of the 15 most frequent inspection observations. FY2018, however, saw changes in the seven observation citations.
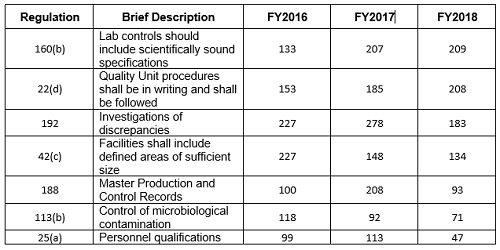
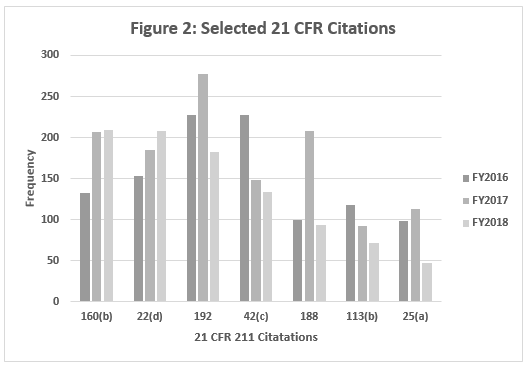
Table 2 and Figure 2 show only the seven citations with notable differences between FY2016 and FY2018. Two regulations increased markedly in the frequency with which they were cited between 2016 and 2018: §211.160(b) (lab controls should include scientifically sound specifications) and §211.22(d) (Quality Unit procedures shall be in writing and shall be followed). These have possibly increased in frequency because of the continued FDA focus on OTC manufacturers, including those outside the U.S. These firms often have rudimentary quality systems and quality units that do not exercise their responsibility or authority. Further, these manufacturers frequently fail to ensure that product is tested and meets specifications prior to release. These two citations have displaced §211.192 (investigation of discrepancies) from the top position, where it held that honor for many years.
Five regulations saw a significant decrease in the frequency with which they were cited between 2016 and 2018. This includes §211.192 (Investigations of discrepancies); §211.42(c) (Facilities shall include defined areas of sufficient size);§211.188 (Master Production and Control Records); §211.113(b) (Control of microbiological contamination); and §211.25(a) (Personnel Qualification). Two of these citations fell precipitously in frequency. The frequencies of citations of §211.188 and §211.25(a) fell by more than half between FY2017 and FY2018. It is interesting that the frequency of citation for §211.113(b) has decreased consistently since FY2015, while the FDA continues to identify sterile drug product manufacture as a high-risk operation, and many warning letters identify violations by this type of manufacturer.
Table 1: Drug GMP Inspections, §211 Citation Frequency
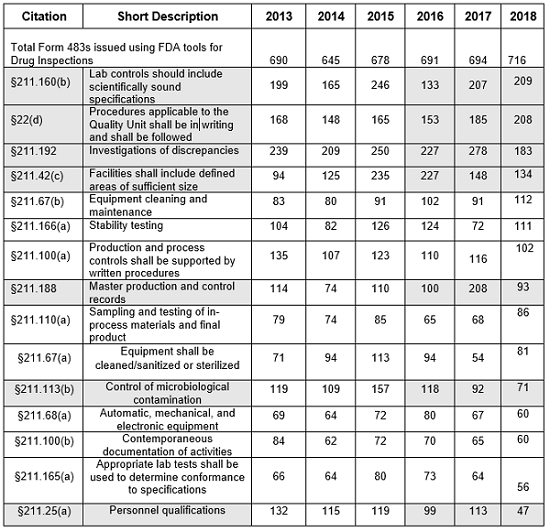
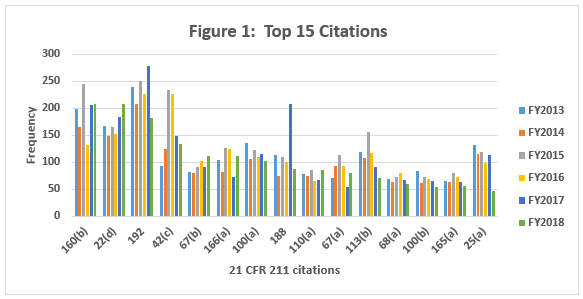
Table 2: FDA Drug Regulation Citations in FY2016 through FY 2018 Inspections for Selected Areas
Conclusions
For those who use inspection observations to monitor and improve their quality systems, the FDA’s annual data provides ample resources against which firms can measure their potential vulnerabilities and gauge the probable focus areas during upcoming GMP inspections.
The dramatic increase in the number of times the FDA cited §211.160(b) between 2016 and 2018 should cause smaller firms, and OTC manufacturers particularly, to ensure they establish scientifically sound specifications and ensure products are tested to meet these prior to distribution. Also, the lack of adequate written procedures and responsibilities for the quality unit, §211.22(d), remains a very consistent citation over the five years addressed herein, with heightened visibility in FY2018. Form 483 observations that include text such as “The Quality Unit is Inadequate…” often result in additional enforcement action, including warning letters.
While §211.192 fell to third place this year and dropped in frequency of citations this year, it nonetheless remains an important observation. It would not be prudent to think investigations were no longer important.
About The Author:
 Barbara Unger formed Unger Consulting, Inc. to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including general GMP auditing and auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Unger was the first chairperson of the Rx-360 Monitoring and Reporting work group that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She is currently the co-lead of the Rx-360 Data Integrity Working Group. You can contact her at bwunger123@gmail.com.
Barbara Unger formed Unger Consulting, Inc. to provide GMP auditing and regulatory intelligence services to the pharmaceutical industry, including general GMP auditing and auditing and remediation in the area of data management and data integrity. Her auditing experience includes leadership of the Amgen corporate GMP audit group for APIs and quality systems. She also developed, implemented, and maintained the GMP regulatory intelligence program for eight years at Amgen. This included surveillance, analysis, and communication of GMP related legislation, regulations, guidance, and industry compliance enforcement trends. Unger was the first chairperson of the Rx-360 Monitoring and Reporting work group that summarized and published relevant GMP and supply chain related laws, regulations, and guidance. She is currently the co-lead of the Rx-360 Data Integrity Working Group. You can contact her at bwunger123@gmail.com.