
How FDA And MHRA Decide Which Drug Facilities To Inspect — And How Often

The FDA recently revamped the methods it uses to determine which foreign and domestic drug manufacturing sites warrant inspection or other types of surveillance and at what frequency. In concert with the method revamp, the agency introduced a multiyear resource planning process that will enable it to better use resources and plan frequencies of product sampling as well as inspections.
At the FDA/Xavier PharmaLink conference at Xavier University in Cincinnati, Ohio, in March, FDA Office of Surveillance (OS) Senior Advisor Beth Philpy discussed the recent changes, why they were enacted, and the impact they will have on the pharmaceutical industry. OS is part of the Center for Drug Evaluation and Research (CDER) Office of Pharmaceutical Quality (OPQ), and it is responsible for coordinating drug inspections. Both offices were created in 2015.
OS was formed by combining different functions from different parts of the agency to enable a “more comprehensive approach” to surveillance, Philpy said. Its focus is on good manufacturing practices (GMPs) and product quality. “We need to know who makes the drugs, where they are coming from, and what products are being made at what sites at any given time,” she explained.
OS is responsible for looking at approximately 7,000 human drug manufacturing sites, about 2,000 of which are medical gas manufacturers. Forty percent of the sites are domestic, and the rest foreign, including 97,000 unique products for finished dosage products and 4,000 unique active pharmaceutical ingredients (APIs), as determined by the materials’ National Drug Codes (NDCs).
Philpy said that the “state of quality” for sites and products is determined by evaluation and trending of internal and external information about regulated sites and products — from submissions to the agency, inspections, product sampling, or open source data (see Figure 1 for a list of data sources used).

Figure 1: FDA Office of Surveillance sources of information
Surveillance Or Inspection?
Philpy explained that OS has both formal and “less formal” processes to determine which companies and which specific plant sites or products may warrant agency attention. “In terms of how we choose who to inspect, it is not just who to inspect, but it is who to surveil in general,” she said. “The on-site inspection is not our only tool for surveillance.”
OS uses a risk-based process to look across the spectrum of firms in its purview and compare manufacturing sites, looking for trends and outliers. Based on the potential risk, it makes decisions on which sites may need inspecting and which may only require product sampling.
Philpy said that while a particular manufacturing site may seem “OK,” the agency may focus on possible issues with certain products or types of products at the site and decide how to engage the firm.
“Maybe there are no incidents of noncompliance, but we are seeing something that may concern us,” Philpy explained. “We can reach out during the course of inspection, or post-inspection, or even surrounding the recent approval of a product, where we can have that slightly less formal engagement to see what may be happening, including what changes may be either occurring or planned.”
In terms of looking at surveillance long-term, she pointed out that “right now our action planning is on an annual basis. But there are things that come up. There are signals that may make us want to look at something on the next inspection or may raise that product’s likelihood of sampling in the following fiscal year. Or there may be a need for more immediate engagement or potential for enforcement. We work closely with the Office of Compliance in that regard.”
Biennial Inspection Model No Longer Applies
In the past, the agency had a goal of inspecting manufacturing sites every two years. The FDA Safety and Innovation Act (FDASIA), signed into law in 2012, supersedes that requirement. All sites are ranked together based on frequency considerations required by FDASIA.
The FDASIA section 705 requirements were built into the CDER-ORA site selection model (SSM). “It is a tool that was developed in 2005,” Philpy said. “It has had changes and additions over the years. It is a risk management tool that we use globally. We use risk-ranking factors in an in-house developed algorithm to prioritize sites on an annual basis.” The agency believes that applying this risk ranking model results in the global parity expectation in FDASIA 705 (see Figure 2).

Figure 2: CDER-ORA site selection model risk-ranking factors
Philpy pointed to Section F — a catch-all section that allows “any other criteria” deemed necessary and appropriate to perform the risk ranking. “We are looking to expand that as best we can to get the most information,” she said. “You can see that a lot of the criteria — for example, A, B, and C — are more negative types of information, from when things go wrong. What we are working on now in addition to making that more robust is looking at the offsets in terms of what the positive things are that we see that can make a better score, to lower the risk for on-site inspection and engagement.”
The model, she said, has three primary sub-scores: facility information and history, product type and history, and time since the last inspection. Each facility and product has numerous other sub-scores. The scores are not absolute, but relative, and act as a prioritization scheme.
Regarding the risk ranking, Philpy said there is a “common misconception” that a site that ranks number one or two is guaranteed to be judged as official action indicated (OAI), i.e., subject to some type of formal enforcement.
“That is not the case at all,” she commented. “It is intended for a two-pronged approach. One is coverage: We do want to cover the entire inventory over a period of time. It is also for prioritizing sites such that if something were to go wrong, that is where it would have the highest impact. It is not that it will go wrong, but if it did, it would be bad — something that we want to avoid.”
Model Continues To Evolve; MRA Input Now Included
The risk ranking site selection model continues to evolve, with algorithms that are dynamic. The most recent iteration included input from experts from the Office of Regulatory Affairs (ORA) — the lead office for all agency field activities. “We engaged new experts to do these rankings of all the various factors involved and put in place a much more documented and structured governance process,” Philpy said.
The newest data source that feeds into the model is the U.S./EU mutual recognition agreement (MRA), which allows sharing of inspection and other information between the FDA and European health authorities, once the individual authorities have been inspected and deemed to have processes that are equivalent to the FDA’s. As of March 2018, 12 European authorities had been found capable. The remaining need to be done in the following 15 months.
“This is new for the FDA,” Philpy said. “Our European partners have all done this before. They have been sharing information with each other for years. So, they had processes in place for how this should work. Our regulatory scheme is a bit different — for example, we do not issue GMP certificates.”
To incorporate the new MRA information, the agency began by reviewing its annual surveillance action plan, focusing on the sites it intended to inspect. It looked at the EudraGMDP database for information about the EU’s inspection schedules and recent outcomes and put that information in its model.
“Again, this is a mutual recognition agreement — we do not fully rely on their information,” Philpy said. “We get the documents in and we review them just as we would an FDA inspection. We are a very evidence-based organization. Should there be anything negative that we need to take forward for enforcement, it is done on an evidentiary basis.”
Leverage Data Already In-House
In efforts to expand its facility and product surveillance and inspection model, the FDA is looking at data that it already has in-house and expanding its cadre of data and risk scientists to evaluate how the data might be used.
Philpy explained that the FDA has “a lot of data that we have not put together or linked. We want to be able to get up to speed and put it all together and use more predictive analytics to be able to look at the information we have.”
For example, a large establishment inspection report (EIR) “is rife with unstructured data. So, we are doing some contextual analysis and asking what we can pull from an EIR that may line up with some recall data or some product quality submission data or anything like that,” Philpy said. Unstructured data is information that either does not have a predefined data model or is not organized in a predefined manner. Unstructured data is typically text-heavy but may contain data such as dates, numbers, and facts as well.
“For us, we can only get the data at certain points — when it is sent to us, when we get it on inspection, or when it is publicly available. To dig and find and match that up to your FEI (FDA Establishment Identifier) or NDC, if we use the ‘crawl, walk, run’ analogy, we are growing up. We may be toddling a little bit. We hope to be able to get there faster by hiring some more risk scientists and data scientists and growing that area significantly. We need to keep the GMP quality mind-set and make sure we have the right context.”
MHRA Inspection Selection Model Also Risk-Based
Also at the Xavier PharmaLink conference, U.K. Medicines and Healthcare products Regulatory Agency (MHRA) Senior GMDP (good manufacturing and distribution practices) Inspector Tracy Moore discussed her agency’s process for determining which manufacturing and importing facilities to inspect, with a focus on how noncompliant inspection findings are escalated at MHRA.
MHRA’s method for determining who to inspect is risk-based, like the FDA’s, and relies on previous inspection findings, from which it assigns a provisional rating. The rating is modified based on a number of other “discriminatory factors” (see Figure 3]).
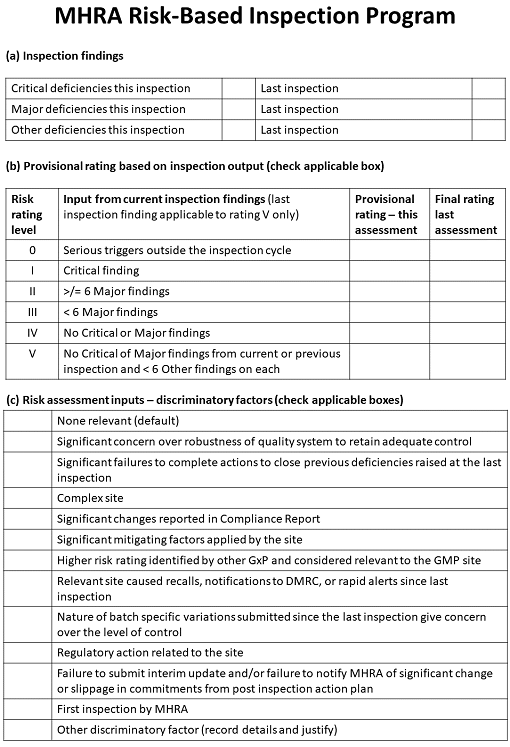
Figure 3: The U.K. Medicines and Healthcare Regulatory products Agency’s risk-based inspection program
This information is combined with inspectors’ input on the factors, resulting in a risk rating for the manufacturing or importing site. The ratings range from 0 — meaning that an inspection should take place as quickly as possible — to 5, meaning that the inspection can wait for 30 months with a 50 percent reduction in the duration of the next inspection. Under this model, all manufacturers and importers should be inspected at least every 30 months.
MHRA Escalation Process Adopted By EMA
Moore detailed the escalation process MHRA uses to deal with issues discovered during a GMP inspection that may require further attention by the agency. The process developed by MHRA is of particular interest as it has now been adopted by the European Medicines Agency (EMA).
“On inspection, when you have been given some quite serious and significant findings, if the inspector feels that something additional needs to happen, but no actual regulatory action needs to occur at that point, this is where the referral to the Compliance Management Team (CMT) comes in,” Moore explained.
The CMT is a group of senior and expert GMDP inspectors with links to the senior inspectorate management. It provides an independent review of inspection findings and responses. When a case has been referred to the CMT, it becomes the point of contact for the company involved.
When a firm is contacted by the CMT and the response it receives is considered inadequate or inappropriate, regulatory action can be escalated up to the Inspection Action Group (IAG). The IAG is a multidisciplinary group that makes recommendations to the MHRA’s director of inspection, enforcement, and standards on regulatory or adverse licensing action. The IAG is made up of senior assessors and inspectors, an attorney from the Department of Health Legal Services, the inspector involved in the inspection under review, a representative from MHRA enforcement, and other senior agency personnel.
As of March 2016, CMT was in the process of reviewing 16 cases, none with critical findings. “We do not expect a CMT case to have a critical finding, because nine times out of 10 those would be an IAG case, because it is the IAG that issues the statements of noncompliance (SONC),” Moore said. A SONC is roughly equivalent to an FDA warning letter.
In the six months prior to Moore’s March 2018 presentation, MHRA issued six SONCs, five of which were related to data falsification. When the IAG takes action or issues a SONC, it is shared with all MRA partners, including the U.S., Japan, Canada, Switzerland, New Zealand, and Australia. Actions that have the potential to be deemed criminal are passed to the enforcement branch. In the U.K., it is a criminal offense to mislead an inspector — an offense that carries a sentence of up to two years in prison.
Moore explained that MHRA does not have to be on-site or do an inspection to issue a SONC. She gave a recent example in which an importing firm was getting failed test results on material that was accompanied by a certificate of analysis (COA) from the manufacturing site stating that the product was in specification. The importation site asked for an MHRA investigation because the qualified person (QP) at the firm refused to certify the material. A SONC was issued based on a comparison of the testing results, which acted like a U.S. FDA import alert, stopping product from being shipped not only into the U.K. but to anywhere in Europe. The site was subsequently inspected, and the SONC remained in place.
About The Author:
 Jerry Chapman is a GMP consultant with nearly 40 years of experience in the pharmaceutical industry. His experience includes numerous positions in development, manufacturing, and quality at the plant, site, and corporate levels. He designed and implemented a comprehensive “GMP intelligence” process at Eli Lilly and again as a consultant at a top-five animal health firm. Chapman served as senior editor at International Pharmaceutical Quality (IPQ) for six years and now consults on GMP intelligence and quality knowledge management and is a freelance writer. You can contact him via email, visit his website, or connect with him on LinkedIn.
Jerry Chapman is a GMP consultant with nearly 40 years of experience in the pharmaceutical industry. His experience includes numerous positions in development, manufacturing, and quality at the plant, site, and corporate levels. He designed and implemented a comprehensive “GMP intelligence” process at Eli Lilly and again as a consultant at a top-five animal health firm. Chapman served as senior editor at International Pharmaceutical Quality (IPQ) for six years and now consults on GMP intelligence and quality knowledge management and is a freelance writer. You can contact him via email, visit his website, or connect with him on LinkedIn.