
Quality Risk Management 101: Risks Associated With Medicinal Products
By Kelly Waldron, Ph.D., ValSource, Inc.
This article marks the first of a series intended to provide a holistic primer on the field of quality risk management (QRM). The series will provide background information for those new to the QRM discipline and will explore topics including the various types of risks associated with medicinal products, the role of risk management in pharmaceutical and biopharmaceutical regulation, QRM principles and practices, the role of QRM across the product life cycle, primary literature sources for QRM, and common challenges associated with QRM implementation.
The series will provide background information for those new to the QRM discipline and will explore topics including the various types of risks associated with medicinal products, the role of risk management in pharmaceutical and biopharmaceutical regulation, QRM principles and practices, the role of QRM across the product life cycle, primary literature sources for QRM, and common challenges associated with QRM implementation.
To establish a quality risk management program and ensure that the appropriate types of risks remain at the forefront of subsequent QRM efforts, it is necessary for QRM practitioners to understand the breadth of risks to which the patient might be exposed. Within the realm of medicinal products, two general categories of risk to the patient can be identified: intrinsic risk and extrinsic risk. Table 1 delineates the various characteristics of, and differences between, these two categories of risk.
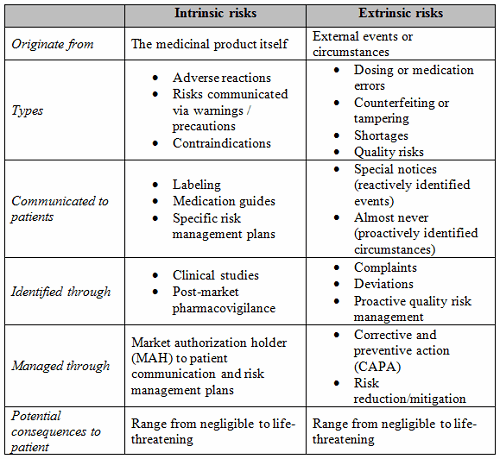
Intrinsic risks are those inherent to a drug, given the nature of certain biochemical reactions within the body when exposed to the drug or its constituent parts. Intrinsic risks generally surface during research and development and in the clinic and are weighed against the overall medical benefits of the product when determining whether the drug is suitable for commercial sale. For example, while the risk of suicidal thoughts or feelings might be considered acceptable in the context of an antidepressant medication where such underlying urges may already be present, that same risk would be unacceptable if it were to accompany a mild pain reliever such as ibuprofen. The acceptability of intrinsic risks is therefore relative, based on the therapeutic benefits a drug delivers. While intrinsic risks are rarely discussed within the realm of commercial pharmaceutical manufacturing and the chemistry, manufacturing, and controls (CMC) portion of the product life cycle, QRM practitioners should possess adequate knowledge of these risks to understand the underlying relationship between these and the quality risks that are the focus of QRM.
Understanding Intrinsic Risks
Generally, intrinsic risks are communicated to patients and their healthcare providers through product labeling so that patients, in consultation with their medical teams, can make informed decisions regarding the course of their care. While requirements for drug product labeling vary by region, in most instances the communication of intrinsic risks can be subdivided into categories, including adverse reactions, warnings and precautions, and contraindications. Adverse reactions (known in some regions as side effects or undesirable effects) are conditions that are undesirable in nature that the patient might reasonably expect to occur during use of the medicinal product.1 These may include (but are certainly not limited to) dry mouth, drowsiness, stomach pain, nausea, vomiting, or diarrhea. Side effects are often discovered over the course of clinical trials and may be further refined through data from adverse event reporting, complaints, and pharmacovigilance activities that occur in the commercial phase of the product life cycle.
Warnings and precautions are extensions of adverse reactions that might threaten the patient’s health and well-being, either because the adverse reaction is severe enough to impact decisions regarding a patient’s care or because the adverse reaction might require more specific precautions to be communicated to the patient.2 For example, the common analgesic and fever reducer paracetamol (called acetaminophen in the U.S.) comes with a warning regarding liver failure in doses in excess of recommendations.3;4 Intrinsic risks grouped within warnings and precautions are often discovered during drug development but may also be included to highlight logical consequences of known side effects (as is the case, for example, when drugs that cite drowsiness as an adverse reaction also include a precaution against driving and operating heavy machinery). Warnings and precautions, therefore, are derived from risk assessments in which the gravity of the risk (likelihood of harm, severity of the harm, or a combination thereof) or consequences of drug administration might pose a threat to the patient in an unintended way.
Contraindications are an additional class of intrinsic risk in which a causal relationship between the drug and some other physiological condition (a disease condition or cohort of the patient population) exists and may result in an adverse reaction.2 For example, certain drugs may be contraindicated for use in children, pregnant or lactating women, or the elderly, while others may be ill-advised for administration when the patient is immunocompromised due to a co-existing condition. In addition, certain drugs may interact with other drugs, resulting in serious consequences that jeopardize the patient’s overall health, as is the case when two classes of antidepressant drugs (monoamine oxidase inhibitors, or MAOIs, and selective serotonin reuptake inhibitors, or SSRIs) are used simultaneously.5
Balancing Intrinsic Risks, Medicinal Benefits
Regulatory authorities around the world base product approval decisions on a variety of factors, the most important of which is the benefit-risk profile (also referred to as “risk-benefit”). This evaluation weighs the intrinsic risks of the product (such as the nature of adverse reactions that may occur, the relative severity of the reactions, and their estimated or confirmed rate of occurrence in the target patient population) against the medical benefits of the product. In most cases, the benefits of the product must clearly outweigh the risks in order to be considered appropriate for commercial use; panels of experts are often used to guide the final decision, though quantitative risk assessment models may be employed to calibrate the scientific judgment of decision makers.6 In some cases, however, the data may reveal that the benefits outweigh the risks only in certain circumstances, or certain risks are severe enough to warrant additional management to ensure the benefit-risk profile remains favorable. In these instances, a regulatory authority may call for the proposal and enactment of a risk management plan (RMP) to manage these risks in a commercial setting.
Risk management plans (referred to as risk evaluation and mitigation strategies, or REMS, in the U.S.) manage intrinsic risk through the application of targeted controls. The nature of the controls varies based on the risk and are intended to assure that the medical benefits of the drug product continue to outweigh the risks.7 For example, the medication isotretinoin (originally marketed under the brand name Accutane) offers significant relief to patients with severe acne, a painful condition that can negatively impact patients’ self-confidence. Isotretinoin is incredibly effective in the treatment of acne, in most cases eliminating the condition altogether; however, it can also cause severe birth defects.8 Despite the fact that the drug’s labeling documents a contraindication for pregnant women, a risk management plan was deemed necessary to ensure that women would not become pregnant while using the product. The REMS for isotretinoin, called iPledge, includes a medication guide, a certification program for prescribing physicians and dispensing pharmacies, and mandatory enrollment of patients in the iPledge program. Risk controls associated with the iPledge program include limited prescribing allowances (no more than 30 days of medication with no refills at any time), contraception counseling between the healthcare provider and patient, two pregnancy tests performed prior to drug administration with monthly follow-up tests thereafter, certification from the patient that she will use two forms of contraception throughout the treatment period, and other similar controls9 These additional risk controls are intended to ensure that the benefits of isotretinoin (the treatment and potential elimination of severe acne) outweigh the risks (severe birth defects) by preventing fetal exposure to the drug.
Regulators and industry acknowledge that the data obtained during drug development and clinical testing may not be sufficient to reveal all intrinsic risks. As a result, ongoing pharmacovigilance is required. Pharmacovigilance is “the science and activities relating to the detection, assessment, understanding and prevention of adverse effects or any other possible drug-related problems.”10 Conducted in a post-market environment, these programs enable the identification of signals that might indicate a new intrinsic risk, or provide additional data regarding the frequency and severity of known risks that can further inform the benefit-risk balance.11 New information gleaned from pharmacovigilance may result in the discontinuation of a product. For example, the popular weight loss drug fenfluramine was discontinued in 1997 after pharmacovigilance activities revealed a correlation with heart valve disease and pulmonary hypertension, conditions not previously identified in clinical trials.12
Historically, pharmacological research and drug approval regulation have focused primarily on these intrinsic risks; as such, intrinsic risks garnered the most industry and regulatory scrutiny. However, intrinsic risks are only a portion of the total risk a patient may be exposed to during their use of a medicinal product. The other category of risks, extrinsic risks, may have similarly grave consequences, yet they have been comparatively neglected until recent times.
Quality Risk Management Focuses On Extrinsic Risks
Extrinsic risks can be defined as unintended risks to the patient arising from events or circumstances unrelated to the drug product itself; for example, dosing errors, counterfeiting or tampering, drug shortages, or risks introduced during manufacturing, packaging, labeling, distribution, or storage. A critical subset of these are quality risks — risks to patients that are associated with the quality of the medicinal product. These are the risks that constitute the focus of quality risk management.
Quality risks are a nefarious bunch. Because quality problems often manifest as failures to meet specifications, and because drug specifications address chemical and biochemical attributes of the product, quality problems are often invisible to the patient. Quality problems can vary by manufacturer and batch — as such, the same drug might expose patients to different quality risks, depending on the brand or product lot number. Patients are rarely informed of the potential quality risks associated with a given product, limiting their ability to make informed choices about their healthcare. Finally, quality risks may greatly affect the benefit-risk balance of product, since products are approved for sale based on a favorable benefit-risk ratio, and quality risks introduce new considerations into that equation.
Patients and healthcare providers make an inherent assumption that a product’s intrinsic risk has been accepted through regulatory approval and that the drug they administer or receive is both safe and effective. Regulators and industry acknowledge, of course, that deviations from cGMP and product-specific quality attributes could threaten the critical link between an individual dose of a medicinal product and its marketing authorization, thereby rendering the drug “adulterated.” In other words, any quality risk imposed in addition to the drug’s intrinsic risk upsets the benefit-risk balance and should therefore be identified and controlled.
When patients take a medicine, they exercise their trust in the pharmaceutical industry. They trust that the product is as it is labeled and that no external circumstance might put them at risk beyond that disclosed in the labeling. They trust that drug manufacturers have produced the product in a safe and consistent way and that their health and safety are protected. The market does not distinguish between high- or low-quality drugs, because acceptable quality is presumed to be present.13
Those familiar with quality-related recalls and GMP-related warning letters understand that this assumption is far from the truth. While the sources of quality problems, consequences of poor quality drug products, and the resultant patient impacts have been well documented, a rigorous inquiry into a methodological approach to the resolution of these issues was notably absent until the advent of the discipline of quality risk management.
The next article in this series offers a brief history of risk management for pharmaceutical products, expanding upon the evolution of the field of QRM in pharmaceuticals and biopharmaceuticals.
References:
- FDA. Guidance for Industry: Adverse Reactions Section of Labeling for Human Prescription Drug and Biological Products - Content and Format. Jan 2006.
- FDA. Guidance for Industry: Warnings and Precautions, Contraindications, and Boxed Warning Sections of Labeling for Human Prescrption Drug and Biological Products - Content and Format. Oct 2011.
- GlaxoSmithKline Consumer Healthcare. Panadol patient leaflet. Medicines.ie. [Online] [Cited: Sep 21, 2016.] http://www.medicines.ie/medicine/16259/PIL/Panadol+500mg+Film+Coated+Tablets/
- Johnson and Johnson Consumer, Inc., McNeil Consumer Healthcare Division. Safety and Dosing Frequently Asked Questions. Tylenol. [Online] [Cited: Sep 21, 2016.] https://www.tylenol.com/safety-dosing/faq#general.
- Dunner, D.L. Combining antidepressants. : Shanghai Archives of Psychiatry, 26 (6). Dec 2014.
- EMA, Committee for Medicinal Products for Human Use. Reflection Paper on Benefit-Risk Assessment Methods in the Context of the Evaluation of Marketing Authorisation Applications for Medicinal Products for Human Use. Mar 2008.
- FDA. A Brief Overview of Risk Evaluation & Mitigation Strategies (REMS). [Online] [Cited: Sep 25, 2016.] http://www.fda.gov/AboutFDA/Transparency/Basics/ucm325201.htm.
- American Academy for Dermatology. Isotretinoin: Treatment for severe acne. Acne and Rosacea. [Online] [Cited: Sep 25, 2016.] www.aad.org/public/diseases/acne-and-rosacea/isotretinoin-treatment-for-severe-acne.
- FDA. Risk Evaluation and Mitigation Strategies (REMS): The iPledge Program - Single Shared System for Isotretinoin. Apr 2012.
- WHO. The Safety of Medicines in Public Health Programmes: Pharmacovigilance an essential tool.
- EC. The EU pharmacovigilance system. [Online] [Cited: Sep 24, 2016.] http://ec.europa.eu/health/human-use/pharmacovigilance/index_en.htm.
- Elliot, W.T. and J. Chan. Fenfluramine and dexfenfluramine withdrawn from market. AHC Media. Nov 1, 1997.
- Woodcock, J. and M. Wosinska. Economic and Technological Drivers of Generic Sterile Injectable Drug Shortages. Nature. February 2013, Vol. 93, 2.
About The Author:
 Kelly Waldron is currently a senior consultant with ValSource and a member of the Pharmaceutical Regulatory Science Team (PRST) at the Dublin Institute of Technology in Dublin, Ireland. She has particular expertise and a specialized focus on the development and implementation of innovative approaches to quality risk management (QRM). Her expertise also extends to various quality functions in the pharmaceutical, biopharmaceutical, and medical device industries, including quality system design, quality strategy and planning, deviations/investigations, CAPA, change management, audit and inspection programs and response, stability programs, and design control. In addition, Waldron has authored numerous industry and academic papers on QRM. She has a BA in biology from Boston University, an MBA in pharmaceutical management from Fairleigh Dickinson University, and a Ph.D. in pharmaceutical regulatory science (thesis in QRM) from the Dublin Institute of Technology. She can be reached at kwaldron@valsource.com.
Kelly Waldron is currently a senior consultant with ValSource and a member of the Pharmaceutical Regulatory Science Team (PRST) at the Dublin Institute of Technology in Dublin, Ireland. She has particular expertise and a specialized focus on the development and implementation of innovative approaches to quality risk management (QRM). Her expertise also extends to various quality functions in the pharmaceutical, biopharmaceutical, and medical device industries, including quality system design, quality strategy and planning, deviations/investigations, CAPA, change management, audit and inspection programs and response, stability programs, and design control. In addition, Waldron has authored numerous industry and academic papers on QRM. She has a BA in biology from Boston University, an MBA in pharmaceutical management from Fairleigh Dickinson University, and a Ph.D. in pharmaceutical regulatory science (thesis in QRM) from the Dublin Institute of Technology. She can be reached at kwaldron@valsource.com.