
Quality Risk Management 101: A Brief History Of Risk Management In The Regulation Of Medicinal Products
By Kelly Waldron, Ph.D., ValSource, Inc.

This article is the second in a series of six articles intended to provide a holistic primer on the field of quality risk management (QRM). The first article, Quality Risk Management 101: Risks Associated With Medicinal Products, discussed the difference between intrinsic and extrinsic risks and clarified the scope of QRM efforts. Future articles will discuss QRM principles and practices, the role of QRM across the product life cycle, primary literature sources for QRM. and common challenges associated with QRM implementation.
Those new to the field of quality risk management (QRM) should be familiar with the history of risk management for pharmaceutical and biopharmaceutical products and the role of risk management as a regulatory tool. This article reviews the origins of pharmaceutical risk management and regulatory thinking that led to the establishment of QRM as a unique discipline in pharmaceutical development and manufacturing.
Early Pharmaceutical Risk Management
Risk management has been a foundational element of the regulation of healthcare products since the inception of related regulatory bodies; indeed, one could argue that the primary reason such regulatory bodies exist is to protect the public from health and safety risks associated with medicinal product use.
Some sources date early formularies, known as pharmacopeias, back to first century AD Greek texts (such as Pliny’s catalogue of medicinal herbs in Naturalis Historae).1 The earliest known regulation for such pharmacopeia was the Salerno Medical Edict issued by Frederick II of Sicily in 1240, which required apothecaries to prepare their medicinal remedies in the same way.2 Such laws, which became increasingly pervasive throughout the European continent during medieval times, recognized that consistency across drug formulations was necessary to assure the intended effects of the product, thereby minimizing risk to the patient.
The late 19th century saw additional drug legislation come into effect. In the U.S., the first such legislation occurred following the Mexican-American War of 1846-1848, during which American soldiers were administered various drugs for a host of maladies (including malaria, yellow fever, and cholera). Many of these drugs were imported, and some proved to lack the safety and efficacy needed to fully protect the troops. The large number of deaths that occurred in that period can be attributed not only to the typical slaughter seen in wartime, but also to these faulty drugs. The U.S. Import Drug Act of 1848 was sanctioned to ensure that imported drugs were subject to purity and quality testing prior to crossing the border.3 The Import Drug Act established a theme for drug regulation the world over — advances in pharmaceutical regulation generally occur as a consequence of tragedy in the public eye, seeking to manage risk to patient safety and health reactively.
In the U.S., which represents the world’s largest population of drug consumers, the growth in both scope and statute of the FDA was borne of several highly publicized tragedies. Figure 1 illustrates this trend for selected early milestones in American drug law.2,3,4,5
This pattern of reactivity, where healthcare disaster is antecedent to advances in regulatory science, continues to the present day. For example, the heparin scandal of 2008 led to many dozens of deaths, followed by a surge in attention to the management of active pharmaceutical ingredients (APIs) and control over the increasingly complex supply chain.6,7 While this reactive process serves to prevent future injury and death, one is left with regret at the prospect that such tragedies could have been anticipated and avoided with the application of the right tools and the right conviction.
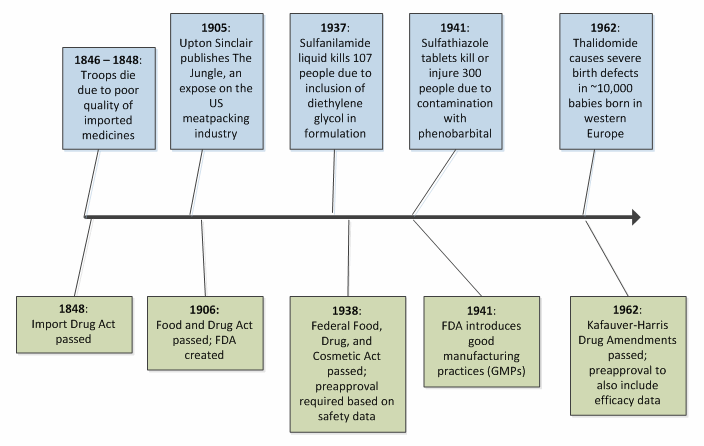
Figure 1: Select timeline of U.S. drug law milestones and public health tragedies
The Precautionary Principle
The precautionary principle represents one of the first proactive risk management mindsets to reach the public sphere. Originally discussed in the context of environmental law, the principle asserts that when faced with uncertainty regarding a given risk, particularly when the consequences of the risk may have serious and lasting effects, an abundance of caution must be used to provide the desired level of protection to society.8,9 The principle serves as a decision-making guideline for regulators, to be invoked in circumstances when scientific evidence regarding a certain risk is lacking. In these cases, a failure to actively avoid the risk could lead to an incredible amount of damage, both of person and of cost; therefore, the only appropriate response is to implement the appropriate measures (such as banning a given substance) to protect the public while simultaneously seeking to increase understanding of the risks.8,9
As a decision aid, the precautionary principle can be viewed as a rudimentary risk management process, as illustrated in Figure 2.8,9,10
The proactive nature of the precautionary principle stems from the early identification of sources of uncertainty, combined with the concerted effort to avoid the associated risk until the uncertainty can be reduced or eliminated. In this way, the concept of risk is linked with scientific knowledge, such that appropriate risk management can only be effectively applied where there is sufficient understanding upon which sound conclusions can be drawn.
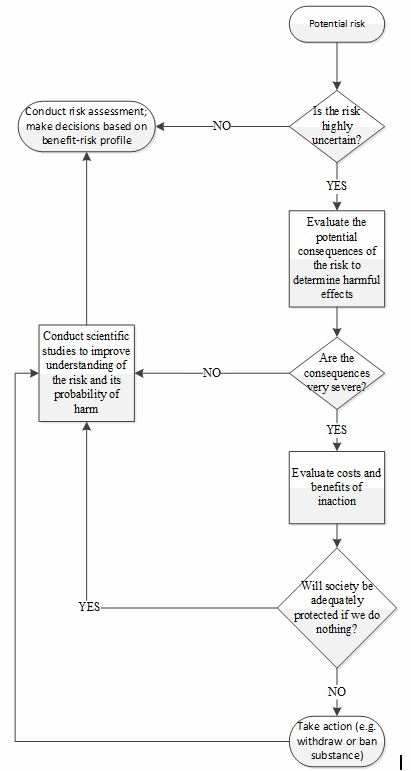
Figure 2: Decision tree illustrating the application of the precautionary principle
Modern Inquiries Into The Role Of Risk Management In Pharmaceutical Regulation
Modern exploration of risk management for drugs and biologics arose with a 1999 report to the FDA commissioner from the Task Force on Risk Management. This task force, established by then-commissioner Dr. Jane Henney, was tasked with determining the technical soundness, consistency, and validity of risk management activities ongoing within the FDA at the time and the development of recommendations to improve the effectiveness and efficiency of these activities. The final report from the task force, Managing the Risks from Medical Product Use: Creating a Risk Management Framework, focused on the premarket benefit-risk assessments performed in support of new drug applications (NDAs) for pharmaceuticals, biological license applications (BLAs) for biopharmaceuticals and biologics, and premarket approvals (PMAs) for medical devices, as well as post-market surveillance activities.11 The report did not explore quality-related risk management, explaining that “injury from product defects is unusual in the United States because of the great attention paid to product quality control and quality assurance during manufacturing.”11 Despite this claim, the report goes on to cite several case studies of injury and death that, through a contemporary understanding of product quality, could be traced to a lack of QRM.
One example describes a spate of product mix-ups that led to the administration of the wrong drug in a hospital setting, leading to three injuries and one death. The distributor, Burroughs Wellcome, packaged the implicated product in a manner similar to other products — including a foil overlay with a transparent window through which the original product labeling could be viewed. The design of this foil overlay allowed for movement of the product within, allowing the product label to slip below the viewing window, rendering the contents of the package difficult to determine. Sadly, this was the root cause of the injuries and death, as the incorrect product was administered to unwitting patients.11 The report did not acknowledge that the application of QRM to the foil overlay design might have allowed for the anticipation and avoidance of such use errors.
Despite the (perhaps myopic) scope of the report, several recommendations were proposed to improve risk communication and early intervention in the event a potential risk is realized.11 These recommendations ultimately contributed to the implementation of several successful programs at the FDA, such as formalization of risk evaluation and mitigation strategies (REMS) and the Sentinel adverse event tracking system, serving the agency’s goal to leverage improved data collection and risk management to better protect public health.
While the 1999 report marked one of the first contemporary explicit inquiries into the existence and effectiveness of risk management and risk-based decision making from regulatory authorities, the topic of quality risk management was not addressed.
A fully formed concept for proactive risk management, including the management of both intrinsic and extrinsic risks, emerged in August 2002 with the announcement of a new FDA initiative titled Pharmaceutical cGMPs for the 21st Century – A Risk-Based Approach. The objectives of this initiative were as follows:
- “Encourage the early adoption of new technological advances by the pharmaceutical industry
- Facilitate industry application of modern quality management techniques, including implementation of quality systems approaches, to all aspects of pharmaceutical production and quality assurance
- Encourage implementation of risk-based approaches that focus both industry and Agency attention on critical areas
- Ensure that regulatory review, compliance, and inspection policies are based on state-of-the-art pharmaceutical science
- Enhance the consistency and coordination of FDA’s drug quality regulatory programs, in part, by further integrating quality systems approaches into the Agency’s business processes and regulatory policies concerning review and inspection activities”12
The final report on the initiative, issued in September 2004, laid out the framework through which the FDA intended to meet or encourage these objectives. While only one of the goals explicitly listed risk management as a focus area, a careful reading of the final report reveals that risk principles underpin the plan.
The report foretold the adoption of a quality systems model for quality management and regulation, to be applied by both industry and the FDA alike. While the quality systems concept had been implemented for some time within medical device regulation (for example, within ISO 13485, Medical devices – quality management systems – requirements for regulatory purposes, and 21 CFR 820, Quality System Regulation), the idea of such a system within pharmaceutical and biopharmaceutical circles was novel.
Several advances in regulatory science had been made under the umbrella of the 21st Century initiative, combining knowledge gained through state-of-the art science and technology with a risk-based orientation. These include, for example:
- Creation of a risk-based model for inspectional oversight
- Issuance of a new guidance on 21 CFR, Part 11, Electronic Records, Electronic Signatures, to encourage the use of risk-based approaches in the adoption of the requirements
- Issuance of a new guidance on aseptic processing, Sterile Drug Products Produced by Aseptic Processing – Current Good Manufacturing Practice, to emphasize the need to proactively prevent contamination during sterile product manufacturing and to further encourage the adoption of risk management principles in the assurance of sterility12
The 21st Century initiative marked a paradigm shift in pharmaceutical regulation: a transition away from rule-based compliance (in which the emphasis was on following statute, often at the expense of developing a deep understanding of products, processes, and associated risks) toward a risk-based view of quality and compliance. In the context of this research, perhaps the most interesting emphasis throughout the 21st Century initiative final report is the repeated use of the phrase efficient risk management. The implications here are, of course, that risk management, if not performed properly, can be inefficient. This is quite a curious prospect, given that one of the reasons a risk-based framework would be employed for a given problem is to ensure that resources are efficiently allocated to the things that matter most. The concept that risk management should be performed in an efficient and effective manner to yield an efficient and effective outcome for the patient is one that, while coveted by industry, remains elusive.
The next article in this series discusses the principles and process of quality risk management based on what is commonly considered the principal governing document in the field, ICH Q9 Quality Risk Management.
References:
- van Tellingen, C. Pliny's pharmacopoeia or the Roman treat. Netherlands Heart Journal, 15 (3). Mar 2007.
- Rago, L. and Santoso, B. Drug Regulation: History, Present and Future. [ed.] C.J. van Boxtel, B. Santoso and I.R. Edwards. Drug Benefits and Risks: International Textbook of Clinical Pharmacology, 2nd edition. s.l. : IOS Press, 2008.
- Ranhalker, H. Historical Overview of Pharmaceutical Industry and Drug Regulatory Affairs. Pharmaceutical Regulatory Affairs. S11-002, 2012.
- FDA. A History of the FDA and Drug Regulation in the United States. [Online] [Cited: Aug 20, 2016.] http://www.fda.gov/centennial/history/history.html.
- Wilkins Parker, J. Risk Management in the United States. [Online] [Cited: Aug 20, 2016.] http://www.fda.gov/downloads/Drugs/ResourcesForYou/HealthProfessionals/UCM473163.pdf.
- Greenemeier, L. Heparin Scare: Deaths from Tainted Blood-Thinner Spur Race for Safe Replacement. Scientific American. [Online] Nov 4, 2008. [Cited: Aug 20, 2016.] http://www.scientificamerican.com/article/heparin-scare-deaths/.
- Pew Health Group. After Heparin: Protecting Consumers from the Risks of Substandard and Counterfeit Drugs. 2011.
- EC. COM(2000) 1. On the precautionary principle. Feb 2000.
- WHO. The precautionary principle: protecting public health, the environment, and the future of our children. 2004.
- United Nations Education, Science, and Cultural Organization (UNESCO), World Commission on the Ethics of Scientific Knowledge and Technology. The Precautionary Principle. Mar 2005.
- FDA. Managing the Risks from Medical Product Use: Creating a Risk Management Framework. May 1999.
- FDA. Pharmaceutical cGMPs for the 21st Century — A Risk Based Approach. Final Report. Sep 2004.
About The Author:
 Kelly Waldron is currently a senior consultant with ValSource and a member of the Pharmaceutical Regulatory Science Team (PRST) at the Dublin Institute of Technology in Dublin, Ireland. She has particular expertise and a specialized focus on the development and implementation of innovative approaches to quality risk management (QRM). Her expertise also extends to various quality functions in the pharmaceutical, biopharmaceutical, and medical device industries, including quality system design, quality strategy and planning, deviations/investigations, CAPA, change management, audit and inspection programs and response, stability programs, and design control. In addition, Waldron has authored numerous industry and academic papers on QRM. She has a BA in biology from Boston University, an MBA in pharmaceutical management from Fairleigh Dickinson University, and a Ph.D. in pharmaceutical regulatory science (thesis in QRM) from the Dublin Institute of Technology. She can be reached at kwaldron@valsource.com.
Kelly Waldron is currently a senior consultant with ValSource and a member of the Pharmaceutical Regulatory Science Team (PRST) at the Dublin Institute of Technology in Dublin, Ireland. She has particular expertise and a specialized focus on the development and implementation of innovative approaches to quality risk management (QRM). Her expertise also extends to various quality functions in the pharmaceutical, biopharmaceutical, and medical device industries, including quality system design, quality strategy and planning, deviations/investigations, CAPA, change management, audit and inspection programs and response, stability programs, and design control. In addition, Waldron has authored numerous industry and academic papers on QRM. She has a BA in biology from Boston University, an MBA in pharmaceutical management from Fairleigh Dickinson University, and a Ph.D. in pharmaceutical regulatory science (thesis in QRM) from the Dublin Institute of Technology. She can be reached at kwaldron@valsource.com.