
Quality Risk Management 101: ICH Q9 In Context
By Kelly Waldron, Ph.D., ValSource, Inc.

This is the third article in a series of six intended to provide a holistic primer on the field of quality risk management (QRM). The first article, Quality Risk Management 101: Risks Associated With Medicinal Products, discussed the difference between intrinsic and extrinsic risks and clarified the scope of QRM efforts and was followed by Quality Risk Management 101: A Brief History Of Risk Management In The Regulation Of Medicinal Products. Future articles will discuss the role of QRM across the product life cycle, primary literature sources for QRM, and common challenges associated with QRM implementation.
International Conference on Harmonisation (ICH) guideline Q9, Quality Risk Management, represents the first internationally recognized guideline specifically addressing QRM for the pharmaceutical and biopharmaceutical industries. Published in June 2005, the guideline offers an overview of general quality risk management principles, an example of a risk management life cycle, discussion around the activities that occur in each life cycle phase, and a list of risk tools and quality system areas to which QRM can be applied.1 This article discusses ICH Q9 in detail, including generally accepted interpretations of the intent and application of the guideline.
In the introduction to the guideline, ICH acknowledges that risk management has been used with much success in other industries, as well as to measure and monitor the intrinsic risk of pharmaceuticals, as discussed in the first article in this series.2 The introduction describes the gap the guideline seeks to fulfill — that of a risk management framework addressing quality risks that could ultimately impact the patient. Rightly so, ICH Q9 positions the patient at the heart of all QRM activities by acknowledging that, despite the diversity of stakeholder interests (e.g., regulators, industry, healthcare providers, etc.), the interests of the patient are paramount. In practice, industry often uses product quality as a surrogate for the patient, since the impacts of quality risks are easier and more scientifically and statistically valid to measure. Provided product quality is defined with an appropriate link to patient, as in quality by design (QbD), the application of such a proxy is fitting.
ICH Q9 moves on to immediately dispel a myth that had taken hold in prior industry and regulatory cultures — the concept of zero risk. In older quality paradigms, drug manufacturers sought to eliminate risk from their products and processes, taking their cue from regulators who implied, through regulatory publications and inspections, that no degree of risk was acceptable. ICH, however, acknowledges that “the manufacturing and use of a drug (medicinal) product, including its components, necessarily entail some degree of risk.” This perspective shifts the industry-regulator conversation from one of absolutes, where quality was a black and white concept of right and wrong, to one focused on balance, where the level of risk is managed to protect product quality and patient safety. The challenge therefore transitions from achieving an esoteric concept of “perfect” quality to understanding what constitutes acceptable risk and striving to achieve that state — perhaps the most significant paradigm shift to occur in the history of drug manufacturing and regulation.
Some other misconceptions regarding risk management are addressed in ICH Q9. For example, many associate risk management with the use of rigorous, detailed tools such as failure modes and effects analysis (FMEA; one of the most common tools employed by industry). However, Q9 is careful to apply the principles of risk management to the practice of risk management itself; using formal or less formal approaches is acceptable, provided the effort is proportionate to the risk of the product, process, or system being assessed. This enables industry to embed risk management in all measures of activities, without the need to undertake a formal, resource-intensive exercise. In addition, Q9 is quite clear that the “appropriate use of quality risk management can facilitate but does not obviate industry’s obligation to comply with regulatory requirements and does not replace appropriate communications between industry and regulators.” Compliance with all applicable laws, of course, is mandatory; risk management may not be used to justify noncompliance or to argue why a specific regulatory requirement need not be fulfilled in a specific instance. Rather, QRM can be used to offer perspectives on how best to comply with statutes and to characterize the aspects of quality that are not specifically associated with compliance.
The benefits of a QRM approach are many, ICH Q9 continues. Better assurance of product quality, for example, may be achieved through the proactive identification and avoidance or minimization of quality risks, as well as the identification of sources of variability in the product and manufacturing process that may be targeted for continuous improvement. The decision-making process can be enhanced, as QRM provides a lens through which scientific data and information can be viewed to better weigh options and understand potential outcomes of a given decision. Finally, QRM can “beneficially affect the extent and level of direct regulatory oversight,” ostensibly by increasing regulators’ trust in a company’s self-awareness through transparency of QRM efforts.
Learn more about how to conduct and implement risk management practices at your firm in the webinar:
Applying Quality Risk Management (QRM) in Manufacturing – A Proactive Approach
Per ICH Q9, the benefits of risk management are to be achieved through the application of a QRM life cycle, an example of which is depicted in Figure 1. The QRM life cycle is an iterative process consisting of four primary phases: risk assessment, risk control, risk review, and risk communication, each of which is facilitated by the application of risk management tools. While ICH Q9 acknowledges that other life cycle models might be used, the majority of the industry has adopted the exact life cycle model described in the guideline.
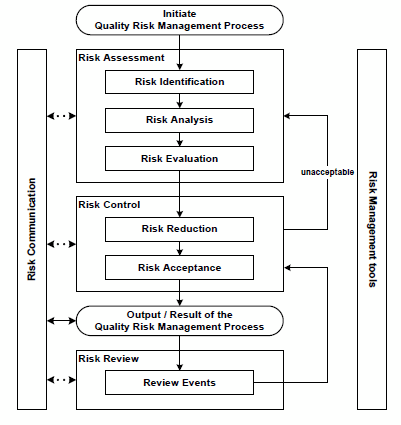
Figure 1: Quality risk management life cycle, per ICH Q9
Unsurprisingly, the first step in the QRM life cycle is the initiation of the process. ICH Q9 describes activities that might be performed during this initiation step, including the identification of resources, leadership, and timelines; specifying the problem statement (also referred to as the risk question); outlining expected deliverables; and gathering applicable data and information that will serve as inputs into the risk management effort. However, Q9 fails to describe when or under what circumstances the QRM process should be initiated; that is, what triggers might exist that should invoke this critical first step.
After initiation, a risk assessment is performed. This phase of the QRM life cycle seeks to determine which risks associated with the product, process, or system under review are unacceptable — a determination made in three general steps. First, hazards are identified as applicable to the problem statement/risk question (risk identification). Each hazard is then analyzed to determine its relative criticality (risk analysis), using the risk equation (likelihood x severity = risk). Finally, the identified and analyzed risks are compared with predefined criteria to determine their acceptability (risk evaluation).1 The risk assessment phase of the QRM life cycle typically draws most heavily upon the use of risk management tools, which allow for a methodical, structured way to identify and analyze risks.
The next phase in the QRM life cycle, risk control, focuses on reducing risks to an acceptable level. This phase is perhaps the most important of the QRM life cycle, as it is the point in the process in which control strategies are identified, implemented, and continuously improved; risk control is the phase that assures adequate protection of the patient. There are two general activities that occur in risk control: the first is a concerted reduction in risk through the application of risk mitigation techniques (risk reduction), and the second includes a confirmation that the risk mitigation actions did not adversely affect the overall risk profile through the introduction of new risks or an increase in risk levels, the risks are adequately controlled (i.e., that the risk mitigation actions and other risk controls are effective), and that the resultant risks are therefore acceptable (risk acceptance). In the event the risk remains unacceptable following risk reduction, the QRM life cycle returns to the risk assessment phase, allowing the practitioner to repeat the process.
Following risk control, there is an output. Though included in the QRM life cycle, ICH Q9 does not devote any narrative description regarding what such an output or result might entail. Typically, this portion of the life cycle is interpreted as a documentation point — the point where the results of the risk assessment and risk control outcomes are drafted into a report that describes the risk assessment outcomes (often formatted to align with the risk management tool employed), risk reduction efforts undertaken, and acceptability of the residual risk.
Once risk control is complete and the results have been documented, the risk review phase of the QRM life cycle begins. The objective of this phase is to ensure that prior activities and associated deliverables remain accurate, relevant, and complete in light of changing conditions. Knowledge gained over the product life cycle, ongoing activities such as changes to the product, process, or system, unplanned events such as deviations and customer complaints, and changes in the internal and external business and regulatory climate have the potential to impact decisions made in the risk assessment and risk acceptance phases of the life cycle. Risk review, therefore, entails a periodic or event-driven review of these changes to determine whether the original risk assessment should be updated (as might be the case when new or previously unrecognized hazards emerge or the original estimates of likelihood and severity have changed) and whether the acceptability of the risk may be affected as a result. In this sense, ICH Q9 positions risk review as an opportunity to confirm the continued validity of decisions made within the QRM process; it does not address a mechanism to determine whether the QRM process (and encompassing program) itself has been effective with respect to reducing risk to the patient.
A critical and often overlooked element of the QRM life cycle is risk communication. Risk communication aims to ensure all applicable stakeholders are aware of risk information, including such aspects as the “existence, nature, form, probability, severity, acceptability, control, treatment, detectability or other aspects of risks to quality.” Such communication most commonly occurs at the output stage of the QRM life cycle, leveraging the documentation associated with the risk assessment and control activities as the primary mechanism to communicate; however, risk communication can, and should, occur at other stages of the QRM life cycle, based on the nature and criticality of the identified risks. Risk communication can occur between varieties of “interested parties,” as depicted in Figure 2.
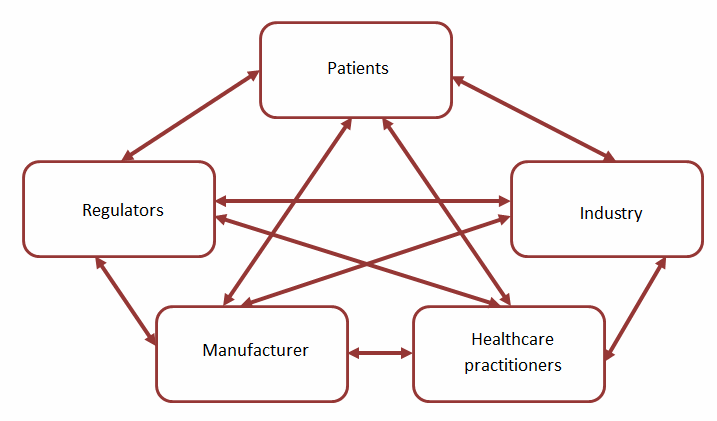
Figure 2: Potential channels for risk communication
A significant challenge in the communication of quality risks lies in the relatively limited options for communication between QRM practitioners and decision makers and the patient. Unlike intrinsic risks (such as known adverse reactions), which are typically communicated through product labeling, extrinsic risks, including quality risks, have no defined mechanism for communication.
ICH Q9 concludes with two annexes: the first describing common risk management tools that may be used to execute the QRM life cycle, and the second describing potential areas for QRM application within the quality system and product life cycle. These annexes are of pivotal importance to the effective implementation of QRM.
Despite ICH’s insistence that the Q9 guideline “is not intended to create any new expectations beyond the current regulatory requirements,” regulatory bodies the world over embraced QRM and have since integrated it at the regional level. Some examples are:
- Inclusion of a new annex to the EU GMPs (Annex 20, which has since been retired and the QRM requirements moved to Chapter 3 of the GMPs), as well as revisions to other directives, annexes, and guidelines to incorporate the principles and practices of quality risk management3
- Inclusion of a new annex to the PIC/S GMP guide (also Annex 20) to adopt ICH Q9 for all member countries4
- Publication of the WHO guidelines on quality risk management5
In this way, QRM has become an integral part of current good manufacturing practices (cGMPs) for pharmaceuticals and biopharmaceuticals, as well as a precision tool to add value to both the pharmaceutical manufacturer and the patient. The next article in this series expands upon this assertion, discussing how QRM has evolved into the foundation of drug development and cGMP platforms, as described in ICH Q8, Q10, and Q11.
References:
- ICH. ICH Q9: Quality Risk Management. Jun 2005.
- Waldron, K. Quality Risk Management 101: Risks Associated with Medicinal Products. Pharmaceutical Online, Jan 2018.
- Eudralex. Annex 20: Quality Risk Management. s.l. : Volume 4, EU Guidelines to Good Manufacturing Practice, Medicinal Products for Human and Veterinary Use, Feb 2008.
- PIC/S. Annex 20, Quality Risk Management. Guide to Good Manufacturing Practice for Medicinal Products, Annexes. Oct 2015.
- WHO. WHO guidelines on quality risk management. WHO Technical Report Series 981, Annex 2. 2013.
About The Author:
 Kelly Waldron is currently a senior consultant with ValSource and a member of the Pharmaceutical Regulatory Science Team (PRST) at the Dublin Institute of Technology in Dublin, Ireland. She has particular expertise and a specialized focus on the development and implementation of innovative approaches to quality risk management (QRM). Her expertise also extends to various quality functions in the pharmaceutical, biopharmaceutical, and medical device industries, including quality system design, quality strategy and planning, deviations/investigations, CAPA, change management, audit and inspection programs and response, stability programs, and design control. In addition, Waldron has authored numerous industry and academic papers on QRM. She has a BA in biology from Boston University, an MBA in pharmaceutical management from Fairleigh Dickinson University, and a Ph.D. in pharmaceutical regulatory science (thesis in QRM) from the Dublin Institute of Technology. She can be reached at kwaldron@valsource.com.
Kelly Waldron is currently a senior consultant with ValSource and a member of the Pharmaceutical Regulatory Science Team (PRST) at the Dublin Institute of Technology in Dublin, Ireland. She has particular expertise and a specialized focus on the development and implementation of innovative approaches to quality risk management (QRM). Her expertise also extends to various quality functions in the pharmaceutical, biopharmaceutical, and medical device industries, including quality system design, quality strategy and planning, deviations/investigations, CAPA, change management, audit and inspection programs and response, stability programs, and design control. In addition, Waldron has authored numerous industry and academic papers on QRM. She has a BA in biology from Boston University, an MBA in pharmaceutical management from Fairleigh Dickinson University, and a Ph.D. in pharmaceutical regulatory science (thesis in QRM) from the Dublin Institute of Technology. She can be reached at kwaldron@valsource.com.