
Quality Risk Management 101: QRM And The Product Life Cycle
By Kelly Waldron, Ph.D., ValSource, Inc.

This is the fourth article in a series of six intended to provide a holistic primer on the field of Quality Risk Management (QRM). The first article, Quality Risk Management 101: Risks Associated With Medicinal Products, discussed the difference between intrinsic and extrinsic risks and clarified the scope of QRM efforts. It was followed by Quality Risk Management 101: A Brief History Of Risk Management In The Regulation of Medicinal Products. The third article in the series, Quality Risk Management 101: ICH Q9 In Context, offered a critical discussion of the QRM process proposed by ICH Q9. The final two articles in the series will discuss primary literature sources for QRM and common challenges associated with QRM implementation.
Quality risk management (QRM) is not a concept to be applied in a vacuum. Rather, QRM is a discipline that provides the most value when used throughout the product life cycle, from the design and development of products, manufacturing process, equipment, and facilities, to the application of the quality system that provides oversight over pharmaceutical production, to the change and life cycle management associated with the product and process. This article reviews each of the areas in turn, providing an overview of the key concepts and how QRM can enhance efforts throughout the product life cycle.
Quality Risk Management And Quality By Design: ICH Q8 And ICH Q11
ICH Q8, Pharmaceutical Development (currently in its second revision), outlines the process for the development of new pharmaceutical and biopharmaceutical products, based on the principles of quality by design (QbD) — a concept originally introduced by Joseph Juran in his 1992 book Juran on Quality by Design: The New Steps for Planning Quality into Goods and Services. QbD, as described in ICH Q8, is considered by many to have brought drug development into the modern era.
In traditional developmental models, a candidate molecule was identified, formulated based on preexisting knowledge of chemistry and pharmacology (or in the case of biopharmaceuticals, standard cell culture and purification processes), scaled to ensure manufacturing processes could serve anticipated demand, and maintained over the commercial life of the product. In many cases, manufacturers, marketing authorization holders (MAHs), and regulators possessed little knowledge of why the product worked and how the associated manufacturing processes supported the clinical effects of the product. As a result, change was demonized; significant efforts were made to keep the manufacturing process static, since the potential implications of change were largely unknown. Over time, this led to antiquated products and associated manufacturing processes, a general reluctance to harness the newest available manufacturing technology, and stalled efforts toward continuous improvement.
In QbD development models, the emphasis is on understanding the linkages between the product and its clinical effects in the patient, the manufacturing process and the product it delivers, and manufacturing systems and the processes they support.1 Quality risk management plays a pivotal role in a QbD development model, by helping to improve the breadth and depth of product and process knowledge and enabling this knowledge to serve as an input into manufacturing process design. The application of QbD (and by extension, QRM) ensures that the manufacturing process delivers a product that consistently meets its specifications, that the defined specifications have meaning in a clinical context.
The first step in applying QbD principles to drug development is to define the quality target product profile (QTPP); that is, the overarching quality characteristics of the product to ensure quality, safety, efficacy, and usability.1 A list of product attributes can be created from the QTPP and preliminary developmental studies. Through the application of QRM principles and tools, the product attributes can be further triaged to identify those that are critical to the patient (critical quality attributes or CQAs) and those that are not. This distinction allows for the focused application of resources (for example, technical and toxicological studies, preclinical and clinical protocol development, and experimental design) to gain knowledge where it matters.
The identified CQAs are used as the input into manufacturing process development and characterization. Given the large number of variables that exist in even the simplest manufacturing process, the need to distinguish between those variables that are critical to ensure the CQAs are met and those that are not becomes clear. Process parameters or variables that have a direct link to CQAs are deemed critical process parameters, or CPPs, and are identified through the application of QRM.1
Manufacturing systems, including facilities, utilities, and equipment, are not discussed in detail in ICH Q8. However, these crucial components are often included in the overarching QbD model, driven by necessity, since manufacturing processes are, by definition, run on equipment, using associated utilities, within a specific-purpose facility. Where a manufacturer chooses to extend its development efforts to manufacturing systems, a similar philosophy applies; the equipment, utilities, and facilities are designed to ensure the CPPs of the associated process are sufficiently controlled.2 The attributes of manufacturing systems that are linked to CPPs are often referred to as critical aspects (CAs).
ICH Q11, Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities), was published in 2012 and reiterates the general concepts of QbD devised in ICH Q8 as applied to drug substances. The novel aspects of ICH Q11, when compared with ICH Q8, surround the discussion of how scientific knowledge and QRM can facilitate intended results in a drug development landscape. ICH Q11 directs attention toward a number of applications of QRM in product and process development, including:
- The evaluation of options for the design of the manufacturing process
- The identification of CQAs and CPPs
- The identification of material attributes (e.g., starting materials, raw materials, reagents, etc.) that may have an impact on CQAs
- The identification of functional relationships that link material attributes and process parameters to the CQAs
- The prioritization of attributes or parameters for further study
- The characterization of how downstream processing could affect the acceptability of risk in upstream unit operations (e.g., how impurities present early in the manufacturing process might be acceptable given the process capability of downstream clearance and purification activities), and
- The definition and continuous improvement of the control strategy and associated monitoring program3
However, based on the relatively limited descriptions of how to employ QRM in ICH Q11, inconsistencies and confusion may result. For example, ICH Q11 notes that “either formal or informal risk management tools… can be used.”3 This statement could violate the tenants of ICH Q9, which notes that the level of formality of QRM should be commensurate with the level of risk. Indeed, it is difficult to envision how the use of an informal risk tool could be sufficient to characterize the critical elements of a product and process and ensure their control as these are perhaps the highest risk aspects of the product life cycle. In addition, ICH Q11 notes that “the risk assessment can also identify CQAs for which there are inherent limitations in detectability in the drug substance (e.g., viral safety). In these cases, such CQAs should be controlled at an appropriate point upstream in the process,” and “when developing a control strategy, a manufacturer can consider implementing controls for a specific CQA at single or multiple locations in the process, depending on the risk associated with the CQA and the ability of individual controls to detect a potential problem.”3 These quotes conflict with the very tenet they seek to explain — that quality cannot be tested into the product, rather it must be built into the product by design. These clauses from ICH Q11 imply that preventive controls should be explored only in the event detection controls are insufficient.
Example 2 of ICH Q11 illustrates the risk ranking of process parameters, indicating how QRM could be used to propose that low-risk parameters be changed without prior regulatory authorization, whereas changes to high-risk parameters would be subject to pre-approval. The use of risk ranking is curious in this context, since it is not the level of risk that would enable this determination but rather the severity of the impact a process parameter might have on a CQA — one-half of the risk equation. For example, a process parameter would be considered critical in the event it exhibits a strong statistical correlation with a given CQA — that is, if variation in the process parameter has been demonstrated to lead to variation in one or more CQAs. However, risk is not a measure of the likelihood that this correlation exists, but rather the likelihood that unacceptable variation will occur, given the control strategy. The ideal state, of course, is that all process parameters deemed critical based on their functional relationship with a given CQA are well-controlled — meaning, have a low probability of affecting a CQA in the context of the process and associated control strategy. The objective is to ensure that all CPPs are likewise low risk.
It is possible that ambiguities of language and imprecision of examples in the regulatory sphere, such as those illustrated above, could result in a reluctance to apply QRM, or worse, incorrect application.
QRM As An Enabler: ICH Q10
ICH Q10, Pharmaceutical Quality System, was published in June 2008 and combined the concepts of QbD, QRM, and GMP into an overarching quality management system to be employed throughout the product life cycle. This document was developed based on existing requirements and guidance, such as ICH Q7, Good Manufacturing Practice Guide for Active Pharmaceutical Ingredients, ISO 9000, Quality Management Systems, and regional GMP requirements.4 ICH Q10 is often grouped with ICH Q8 and ICH Q9 when describing the fundamental paradigm shift that occurred in the pharmaceutical and biopharmaceutical industries in the late 2000s.
The objectives of ICH Q10 are to:
- Achieve product realization, including the definition and attainment of quality specifications as appropriate for the product and patient
- Establish and maintain a state of control, including the definition of what this state of control entails
- Facilitate continual improvement by enhancing the level of control and knowledge of the behaviors of the product and process interface4
In order to facilitate these goals, ICH Q10 reintroduced industry to the product life cycle and described how four primary quality system elements (process performance and product quality monitoring, CAPA, change management, and management review) should be employed over the various life cycle stages. A sizable emphasis is placed on management responsibilities, including ultimate accountability for the quality system and its effectiveness. In addition, ICH Q10 squarely positioned QRM and knowledge management as the two enablers of the quality system. Figure 1 illustrates the product life cycle and the elements of the quality system.
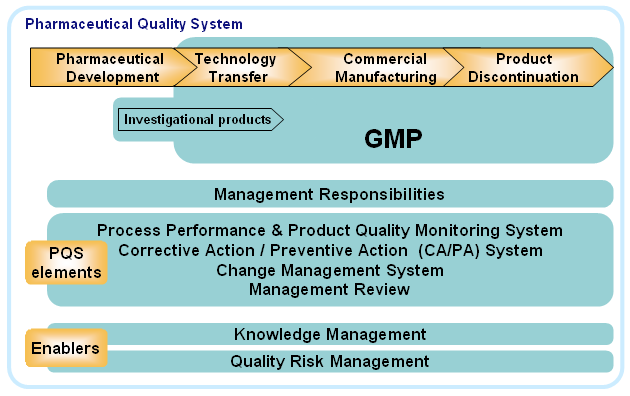
Figure 1: The pharmaceutical quality system5
The product life cycle begins with drug development, a topic discussed in detail in ICH Q8. Following technology transfer, the product is manufactured at commercial scale for sale — products spend the majority of their life cycle in this stage. The final stage of the life cycle is product discontinuation, which may be done for any number of (usually business-driven) reasons, such as the expiration of patent protection or the commercial availability of newer products for the target disease indication. Quality risk management, knowledge management, and the four primary quality system elements facilitate all aspects of the product life cycle, from development through discontinuation.
Risk management principles and practices are embedded in all aspects of the quality system, as illustrated in Table 1. The intersection of each quality system element (column) and product life cycle stage (row) identifies the benefits of applying QRM at that stage.
Table 1: Matrix Illustrating the Primary Functions of QRM at the Intersection of Quality System Elements and Product Life Cycle Stages
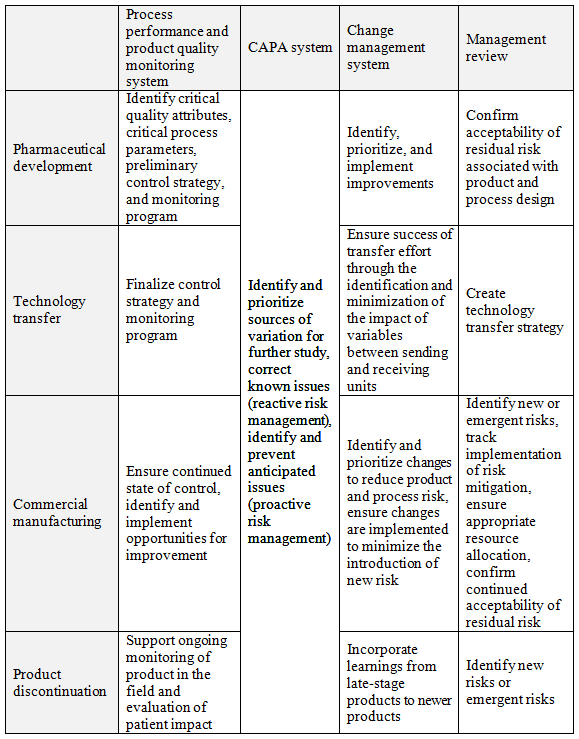
As an enabler of the quality system, QRM is the “engine” that drives the quality system over the product life cycle, transforming information into knowledge and facilitating the identification of opportunities to improve the product and associated processes to better serve the patient.
Quality Risk Management And Life Cycle Management: ICH Q12
In September 2014, ICH announced a new effort to expand the portfolio of quality guidelines with the addition of ICH Q12, Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management. In the final concept paper for this guideline, ICH acknowledged that, true its original name of the International Conference on the Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, the majority of deliverables had focused on premarket requirements. There was a clear need, however, to extend the scope of the group into later stages of the product life cycle, since “the envisioned post-approval ‘operational flexibility’ [outlined in ICH Q8 through Q11] has not been achieved.”6 In order to better facilitate this objective, the Q12 guideline will attempt to:
“…provide a framework to facilitate the management of post-approval Chemistry, Manufacturing and Controls (CMC) changes in a more predictable and efficient manner across the product life cycle. Adoption of this guideline will promote innovation and continual improvement, and strengthen quality assurance and reliable supply of product, including proactive planning of supply chain adjustments. It will allow regulators (assessors and inspectors) to better understand, and have more confidence and trust in, a firm’s Pharmaceutical Quality System (PQS) for management of post-approval CMC changes.”6
ICH later changed its name to the International Council on Harmonization to better align with the newly envisioned scope of influence.7
The announcement of this effort was lauded by industry, as the challenges associated with regional differences in post-approval change management are many.8 Each country has a distinct set of requirements and expectations regarding how such changes may be handled, whether through a firm’s change management program within its quality system (with post-implementation communication to regulators), or through rigorous pre-implementation regulatory approval. This poses logistical difficulties, as companies must juggle inventory manufactured with different variations of change, directing product to specific markets based on the CMC approval status of the applicable regulatory authority.9 This often requires firms to continue manufacturing product under an older manufacturing scheme to ensure consistent supply of product to patients under the jurisdiction of regulatory bodies requiring pre-implementation approval of the change.10 Because of this, many have noted that the global complexity associated with manufacturing change has discouraged innovation and continual improvement.
Among other benefits, ICH Q12 is expected to more clearly link change management with QRM and knowledge management, the two enablers of the quality system as described in ICH Q10.11 In addition, minimization of the regulatory hurdles associated with product and process life cycle management and post-approval change management should spur renewed focus on improvement and innovation.
As discussed throughout this article, modern concepts of cGMP, including quality by design, the pharmaceutical quality system, and effective post-approval change management cannot be fully realized without the appropriate use of QRM. The next article in this series reviews publications that have advanced the discipline of QRM, allowing practitioners to advance their knowledge of the topic by building upon the understanding of experts.
References:
- ICH. ICH Q8(R2): Pharmaceutical Development. Aug 2009.
- ASTM. E2500-07. Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment. Jul 2007.
- ICH. ICH Q11: Development and Manufacture of Drug Substances (Chemical Entities and Biotechnological/Biological Entities). May 2012.
- ICH. ICH Q10: Pharmaceutical Quality System. Jun 2008.
- ICH. How ICH Q8, Q9, Q10 guidelines are working together throughout the product lifecycle. Nov 2010.
- ICH. Final concept paper - Q12: Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management. Jul 2014.
- ICH. Organizational Changes. About ICH. [Online] [Cited: Aug 30, 2016.] http://www.ich.org/about/organisational-changes.html.
- Cox, B. The Post-Approval Challenge of Global Regulatory Complexity. The Gold Sheet. Sep 2014.
- Avenatti, J. Managing Post-Approval Changes in a Global Environment: The Case for Product-Based Impact Assessments. PDA Letter. Jan 2016.
- Garguilo, L. Harmonized Post-Approval Changes: A Vaccine for Global Drug Shortages. Life Science Leader. Mar 2015.
- ICH. Final business plan - Q12: Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management. Jul 2014.
About The Author:
 Kelly Waldron is currently a senior consultant with ValSource and a member of the Pharmaceutical Regulatory Science Team (PRST) at the Dublin Institute of Technology in Dublin, Ireland. She has particular expertise and a specialized focus on the development and implementation of innovative approaches to quality risk management (QRM). Her expertise also extends to various quality functions in the pharmaceutical, biopharmaceutical, and medical device industries, including quality system design, quality strategy and planning, deviations/investigations, CAPA, change management, audit and inspection programs and response, stability programs, and design control. In addition, Waldron has authored numerous industry and academic papers on QRM. She has a BA in biology from Boston University, an MBA in pharmaceutical management from Fairleigh Dickinson University, and a Ph.D. in pharmaceutical regulatory science (thesis in QRM) from the Dublin Institute of Technology. She can be reached at kwaldron@valsource.com.
Kelly Waldron is currently a senior consultant with ValSource and a member of the Pharmaceutical Regulatory Science Team (PRST) at the Dublin Institute of Technology in Dublin, Ireland. She has particular expertise and a specialized focus on the development and implementation of innovative approaches to quality risk management (QRM). Her expertise also extends to various quality functions in the pharmaceutical, biopharmaceutical, and medical device industries, including quality system design, quality strategy and planning, deviations/investigations, CAPA, change management, audit and inspection programs and response, stability programs, and design control. In addition, Waldron has authored numerous industry and academic papers on QRM. She has a BA in biology from Boston University, an MBA in pharmaceutical management from Fairleigh Dickinson University, and a Ph.D. in pharmaceutical regulatory science (thesis in QRM) from the Dublin Institute of Technology. She can be reached at kwaldron@valsource.com.