
7 Steps To Properly Navigate An Event Investigation
By Judy Carmody, Ph.D., Carmody Quality Solutions, LLC

A recently published article examining recent GMP inspection data from CDER (FDA’s Center for Drug Evaluation and Research) and MHRA (Medicines and Healthcare products Regulatory Agency) notes that “Deficiencies in investigations remains at the top of this list [of the most frequently cited observations] over the past four years. We as an industry cannot seem to get this quite right.” I agree. Here’s why.

During a recent event investigation (EI) for a client, I found that, much to my surprise, there was no formal or specific EI process in place to help guide the scientists in the room through the necessary steps. As a result, it was challenging for them to determine how best to approach the investigation at hand. Furthermore, once we started to outline the path forward, there were many different opinions and ideas about how to proceed; the EI bogged down because they were unable to come to a consensus on a plan.
As I observed my colleagues, spoke with them, and collected data, it became apparent that no EI standard operating procedure (SOP) existed. The EI process was combined within a corrective action and preventive action (CAPA) or some other procedure that was too complex and convoluted to follow. They lacked a step-by-step procedure that explained how to conduct an EI.
I am a proponent of SOPs and believe that time spent developing them properly from the start will save much time and effort later. Just as important as having a separate procedure for EI is providing the training necessary to explain an EI, what additional tools are part of this process, and how these tools work together as part of the investigative process under a proper compliance system. I define a “proper compliance system” as one containing event investigations, CAPAs, unplanned deviations, planned deviations, complaints, and risk assessment procedures.
All information is good information during an EI. It is important that the EI is not performed in a bubble — that it is completed in a safe, blameless environment where the community follows specific steps and procedures, and is focused on finding a root cause of a problem rather than conducting a witch hunt. Structuring your compliance system with a separate EI procedure is highly recommended for three reasons:
- It standardizes the process for performing an EI, eliminating inconsistencies in execution and documentation.
- The process is captured in one procedure instead of several procedures, resulting in a more streamlined management of changes or improvements to the process — one change to one document is required, instead of one change to several documents.
- It eases the training burden and promotes greater compliance because it allows for dedicated focus and specificity on an important procedure that will be used by many employees.
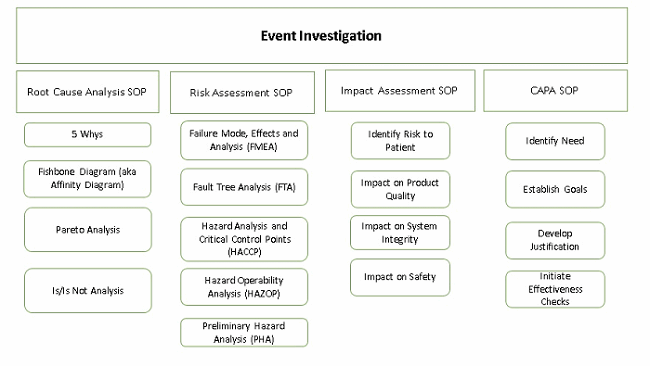
The EI procedure is the first one employees go to when something out of the ordinary, an event or happening, occurs. It is the hub that points to the other procedures (CAPA, risk assessment, etc.) to capture other elements of the investigation. It is used to capture investigations into events including but not limited to complaints, unplanned deviations, unexpected occurrences and confirmed out-of-specification (OOS) results.
Navigating An Event Investigation
Follow these seven steps to properly implement and document event investigations.
Step 1: Describe the Event — Clearly And Concisely
A description of the event includes event details — the what, where, and when. It is stated simply and factually. It is comprised of two or three sentences and does not contain any proposed theories or conclusions, for example, “On 21 Jun 2017 during the manufacture of Vanilla API Lot XYZ123, in manufacturing suite 401, it was observed that the material was clumping and not passing through the sieve on shaker machine #ABC1234.”
Step 2: Collect Data
Data collection is critical to determining the root cause. This step must be done objectively so that all possible data is collected and none is overlooked. Performing this without bias affords the best possibility of properly determining the most likely root cause. Data that should be collected will differ depending on the event but may include: part numbers, lot numbers, environmental conditions, names of all individuals involved, training records, batch records, notebooks, or any labeled or measured material connected with the event.
Step 3: Perform Root Cause Analysis (RCA)
Once the data is collected, a root cause analysis is performed. There are several approaches that can be taken to perform a RCA. There are books written about how to perform RCAs but one approach I find particularly effective and simple to implement is the 5 Whys. An article by John A. Bermingham, a leading drug development and biotech executive, clearly shows not only how to implement it, but also the benefits of this approach.
In cases where a root cause eludes you, it is important to document any probable causes along with a justification for why a root cause was undetermined. This information can inform additional examinations for a root cause.
Step 4: Perform Impact And Risk Assessments
Once the root cause (or possible root cause) is determined, perform impact and risk assessments.
The impact assessment considers the impact of the event on the product or quality system and how it relates to other products or quality systems. To accomplish an impact assessment properly, you must have identified the root cause or have a good idea what it is. Gathering additional data may be required to adequately assess the scope of the impact.
Determination of the root cause (or possible root cause) also serves as the starting point for performing the risk assessment. Many approaches and tools can be used to assess and manage risk, such as:
- Failure mode and effects analysis (FMEA)
- Fault tree analysis (FTA)
- Hazard analysis and critical control points (HACCP)
- Hazard operability analysis (HAZOP)
- Preliminary hazard analysis (PHA)
I am partial to the FMEA approach because it presents as a simple and intuitive process. Usually risk assessments are performed according to a separate procedure and the documentation attached to the governing EI documentation. To perform an FMEA risk assessment, first identify the risks, which consists of the identification of hazards, and the analysis and evaluation of risks associated with exposure to those hazards. Risk identification addresses the following questions:
- What might go wrong?
- What is the likelihood it will go wrong?
- What are the consequences (severity)?
Next, rank risks by severity, likelihood, and detectability. To do this, identify the corresponding risk severity level as high, medium, or low, then determine the likelihood of occurrence of the risk as very likely, likely, or not likely.
Determine the probability of detection by classifying the risk as easily found, somewhat hard to find, or hard to find. Given the severity, risk frequency level, and probability of detection, determine the total risk level. Once the total risk level is determined, the assessment team identifies which remediation strategy is best and designs specific actions (CAPAs) to implement the agreed-upon strategy. Ensure your risk assessments are in keeping with the most current cGMP guidance, which you can find here.
Step 5: Determine CAPAs And Document Changes
Corrective actions (CAs) are subsequent activities taken to eliminate the cause or causes that led to the event, based on what you learned during an event investigation. Preventative actions (PAs) differ from CAs in that PAs are standalone activities, not necessarily related to an event, performed to eliminate the cause(s) of potential nonconformity or other potentially undesirable situations to prevent occurrence. CAPAs determined during the investigation should be executed, documented, and managed according to a separate procedure. One of the benefits of doing this is so the investigation can be closed while the ensuing action(s) are implemented. CAPA documentation is then attached to the governing event investigation documentation. CAPAs include a description of goals, actions to be taken, and evidence and documentation of completion.
Step 6: Form A Conclusion
The conclusion summarizes the event, root cause (or proposed root cause), impact, and risk assessments with reference to any corrective or preventive actions.
The ICH E6 GCP update expects that you have an effective system in place for investigating events. Learn more in the webinar:
CAPA and Root Cause Analysis: Essentials Post ICH GCP E6 (R2) Addendum
Step 7: Initiate Effectiveness Checks (ECs)
If CAPAs are required, effectiveness checks need to be performed. The purpose of ECs is to develop internal monitoring that will generate metrics to evaluate how successful event investigations are managed and if the CAPAs adequately addressed the root cause of the event. This can include:
- Evaluating whether similar events have occurred over a period of time
- What EIs exist and what was done to prevent them
- How many EIs were due to lack of training or proper training
An Event Investigation Case Study
How does all this work in practice? Let’s look at a recent event involving a company that was manufacturing a drug product for a Phase 3 trial. The unexpected event involved identification of a “unknown item” in a sterile reagent. Our investigation set out to collect a range of data, including environmental and raw materials data. We reviewed the batch records to ensure all processes were followed and instrument settings were correct, we examined training records, and we reviewed the same records and data for other batches to determine if there were any differences between the two.
Based on our review of the collected data, we hypothesized that the unknown item in the reagent was microbiological in nature. To verify the hypothesis, we obtained the implicated bottle of reagent and had it tested. The test came back positive. Next, we needed to determine the root cause of the contamination, because just knowing that the item in the reagent was microbiological was not a complete determination of the root cause — we had to find the source of the contamination. Using the 5 Whys approach, we were able to pinpoint several potential sources, including items used during manufacturing and some process-related steps.
During the impact assessment, evaluation of reagent batches manufactured using the same process steps and consumables demonstrated that batches from two different reagents were impacted. A complete review of the consumables used and the process steps was performed to identify corrective actions to be implemented. From this review, several corrective actions were identified and opened. After the corrective actions were implemented, effectiveness checks were performed on batches manufactured subsequent to the changes, verifying that the changes to the process steps and consumables actually eliminated the source of the contamination.
Conclusion
Unexpected events happen — they are a fact of the drug development process. When a company conducts a well-documented event investigation, it informs the process of moving a therapy through development, making formulation or process changes, revising internal documentation, changing packaging configurations, or helping any other host of critical decisions. It can be one of the most important documents a company generates as it provides the rationale and thought process for the decisions made.
All of this goes a long way to help tell the story to auditors so they see the work you have done to deal with the issue. More to the point, it is good science and compliance.
About The Author:
 Judy Carmody, Ph.D., is the founder and principal consultant of Carmody Quality Solutions, LLC, based in the Boston area. With 20+ years of expertise in operations, quality assurance, control, systems, validation, and analytical development, she has a reputation as a quality turnaround specialist in the pharmaceutical and biotechnology industries. Find her on LinkedIn.
Judy Carmody, Ph.D., is the founder and principal consultant of Carmody Quality Solutions, LLC, based in the Boston area. With 20+ years of expertise in operations, quality assurance, control, systems, validation, and analytical development, she has a reputation as a quality turnaround specialist in the pharmaceutical and biotechnology industries. Find her on LinkedIn.